Publications
Publications in peer reviewed journals
Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
2024 - ISME Commun., 4: ycad017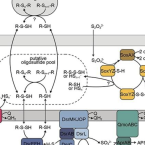
Abstract:
The most abundant known nitrite-oxidizing bacteria in the marine water column belong to the phylum Nitrospinota. Despite their importance in marine nitrogen cycling and primary production, there are only few cultured representatives that all belong to the class Nitrospinia. Moreover, although Nitrospinota were traditionally thought to be restricted to marine environments, metagenome-assembled genomes have also been recovered from groundwater. Over the recent years, metagenomic sequencing has led to the discovery of several novel classes of Nitrospinota (UBA9942, UBA7883, 2-12-FULL-45-22, JACRGO01, JADGAW01), which remain uncultivated and have not been analyzed in detail. Here, we analyzed a nonredundant set of 98 Nitrospinota genomes with focus on these understudied Nitrospinota classes and compared their metabolic profiles to get insights into their potential role in biogeochemical element cycling. Based on phylogenomic analysis and average amino acid identities, the highly diverse phylum Nitrospinota could be divided into at least 33 different genera, partly with quite distinct metabolic capacities. Our analysis shows that not all Nitrospinota are nitrite oxidizers and that members of this phylum have the genomic potential to use sulfide and hydrogen for energy conservation. This study expands our knowledge of the phylogeny and potential ecophysiology of the phylum Nitrospinota and offers new avenues for the isolation and cultivation of these elusive bacteria.
Viral potential to modulate microbial methane metabolism varies by habitat.
2024 - Nat Commun, 1: 1857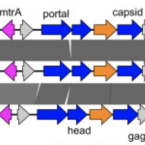
Abstract:
Methane is a potent greenhouse gas contributing to global warming. Microorganisms largely drive the biogeochemical cycling of methane, yet little is known about viral contributions to methane metabolism (MM). We analyzed 982 publicly available metagenomes from host-associated and environmental habitats containing microbial MM genes, expanding the known MM auxiliary metabolic genes (AMGs) from three to 24, including seven genes exclusive to MM pathways. These AMGs are recovered on 911 viral contigs predicted to infect 14 prokaryotic phyla including Halobacteriota, Methanobacteriota, and Thermoproteota. Of those 24, most were encoded by viruses from rumen (16/24), with substantially fewer by viruses from environmental habitats (0-7/24). To search for additional MM AMGs from an environmental habitat, we generate metagenomes from methane-rich sediments in Vrana Lake, Croatia. Therein, we find diverse viral communities, with most viruses predicted to infect methanogens and methanotrophs and some encoding 13 AMGs that can modulate host metabolisms. However, none of these AMGs directly participate in MM pathways. Together these findings suggest that the extent to which viruses use AMGs to modulate host metabolic processes (e.g., MM) varies depending on the ecological properties of the habitat in which they dwell and is not always predictable by habitat biogeochemical properties.
The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
2024 - Acta Obstet Gynecol Scand, in press
Abstract:
Changes within the maternal microbiome during the last trimester of pregnancy and the determinants of the subsequent neonatal microbiome establishment after delivery by elective cesarean section are described.
Maternal vaginal and rectal microbiome samples were collected in the last trimester and before cesarean section; intrauterine cavity, placenta, neonatal buccal mucosa, skin, and meconium samples were obtained at birth; neonatal sample collection was repeated 2-3 days postnatally. Microbial community composition was analyzed by 16S rRNA gene amplicon sequencing. Relative abundance measurements of amplicon sequencing variants and sum counts at higher taxonomic levels were compared to test for significant overlap or differences in microbial community compositions.
gov ID: NCT04489056.
A total of 30 mothers and their neonates were included with available microbiome samples for all maternal, intrauterine cavity and placenta samples, as well as for 18 of 30 neonates. The composition of maternal vaginal and rectal microbiomes during the last trimester of healthy pregnancies did not significantly change (permutational multivariate analysis of variance [PERMANOVA], p > 0.05). No robust microbial signature was detected in the intrauterine cavity, placenta, neonatal buccal mucosa, skin swabs, or meconium samples collected at birth. After birth, the neonatal microbiome was rapidly established, and significantly different microbial communities were detectable 2-3 days postnatally in neonate buccal mucosa and stool samples (PERMANOVA, p < 0.01).
Maternal vaginal and rectal microbiomes in healthy pregnancies remain stable during the third trimester. No microbial colonization of the neonate was observed before birth in healthy pregnancies. Neonatal microbiomes in infants delivered by cesarean section displayed a taxonomic composition distinct from maternal vaginal and rectal microbiomes at birth, indicating that postnatal exposure to the extrauterine environment is the driving source of initial neonatal microbiome development in this cohort.Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
2024 - Microbiome, 1: 55
Abstract:
Microorganisms are responsible for nutrient removal and resource recovery in wastewater treatment plants (WWTPs), and their diversity is often studied by 16S rRNA gene amplicon sequencing. However, this approach underestimates the abundance and diversity of Patescibacteria due to the low coverage of commonly used PCR primers for this highly divergent bacterial phylum. Therefore, our current understanding of the global diversity, distribution, and ecological role of Patescibacteria in WWTPs is very incomplete. This is particularly relevant as Patescibacteria are considered to be associated with microbial host cells and can therefore influence the abundance and temporal variability of other microbial groups that are important for WWTP functioning.
Here, we evaluated the in silico coverage of widely used 16S rRNA gene-targeted primer pairs and redesigned a primer pair targeting the V4 region of bacterial and archaeal 16S rRNA genes to expand its coverage for Patescibacteria. We then experimentally evaluated and compared the performance of the original and modified V4-targeted primers on 565 WWTP samples from the MiDAS global sample collection. Using the modified primer pair, the percentage of ASVs classified as Patescibacteria increased from 5.9 to 23.8%, and the number of detected patescibacterial genera increased from 560 to 1576, while the detected diversity of the remaining microbial community remained similar. Due to this significantly improved coverage of Patescibacteria, we identified 23 core genera of Patescibacteria in WWTPs and described the global distribution pattern of these unusual microbes in these systems. Finally, correlation network analysis revealed potential host organisms that might be associated with Patescibacteria in WWTPs. Interestingly, strong indications were found for an association between Patescibacteria of the Saccharimonadia and globally abundant polyphosphate-accumulating organisms of the genus Ca. Phosphoribacter.
Our study (i) provides an improved 16S rRNA gene V4 region-targeted amplicon primer pair inclusive of Patescibacteria with little impact on the detection of other taxa, (ii) reveals the diversity and distribution patterns of Patescibacteria in WWTPs on a global scale, and (iii) provides new insights into the ecological role and potential hosts of Patescibacteria in WWTPs. Video Abstract.Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
2024 - Environ Sci Technol, 5: 2236-2246
Abstract:
Mycotoxins are toxic chemicals that adversely affect human health. Here, we assessed the influence of mycotoxin exposure on the longitudinal development of early life intestinal microbiota of Nigerian neonates and infants (NIs). Human biomonitoring assays based on liquid chromatography tandem mass spectrometry were applied to quantify mycotoxins in breast milk ( = 68) consumed by the NIs, their stool ( = 82), and urine samples ( = 15), which were collected longitudinally from month 1-18 postdelivery. Microbial community composition was characterized by 16S rRNA gene amplicon sequencing of stool samples and was correlated to mycotoxin exposure patterns. Fumonisin B (FB), FB, and alternariol monomethyl ether (AME) were frequently quantified in stool samples between months 6 and 18. Aflatoxin M (AFM), AME, and citrinin were quantified in breast milk samples at low concentrations. AFM, FB, and ochratoxin A were quantified in urine samples at relatively high concentrations. and / were dominant in very early life stool samples (month 1), whereas was dominant between months 3 and 6. The total mycotoxin levels in stool were significantly associated with NIs' gut microbiome composition (PERMANOVA, < 0.05). However, no significant correlation was observed between specific microbiota and the detection of certain mycotoxins. Albeit a small cohort, this study demonstrates that mycotoxins may influence early life gut microbiome composition.
Reevaluation and novel insights into amino sugar and neutral sugar necromass biomarkers in archaea, bacteria, fungi, and plants
2024 - Sci. Total Environ, 906: 167463
Abstract:
Soil microbial necromass is an important contributor to soil organic matter (>50%) and it is largely composed of microbial residues. In soils, fragmented cell wall residues are mostly found in their polysaccharide forms of fungal chitin and bacterial peptidoglycan. Microbial necromass biomarkers, particularly amino sugars (AS) such as glucosamine (GlcN) and muramic acid (MurA) have been used to trace fungal and bacterial residues in soils, and to distinguish carbon (C) found in microbial residues from non-microbial organic C. Neutral sugars (NS), particularly the hexose/pentose ratio, have also been proposed as tracers of plant polysaccharides in soils. In our study, we extended the range of biomarkers to include AS and NS compounds in the biomass of 120 species belonging to archaea, bacteria, fungi, or plants. GlcN was the most common AS found in all taxa, contributing 42–91% to total AS content, while glucose was the most common NS found, contributing 56–79% to total NS. We identified talosaminuronic acid, found in archaeal pseudopeptidoglycan, as a new potential biomarker specific for Euryarchaeota. We compared the variability of these compounds between the different taxonomic groups using multivariate approaches, such as non-metric multidimensional scaling (NMDS) and partial least squares discriminant analysis (PLS-DA) and statistically evaluated their biomarker potential via indicator species analysis. Both NMDS and PLS-DA showcased the variability in the AS and NS contents between the different taxonomic groups, highlighting their potential as necromass residue biomarkers and allowing their extension from separating bacterial and fungal necromass to separating microbes from plants. Finally, we estimated new conversion factors where fungal GlcN is converted to fungal C by multiplying by 10 and MurA is converted to bacterial C by multiplying by 54. Conversion factors for talosaminuronic acid and galactosamine are also proposed to allow estimation of archaeal or all-microbial necromass residue C, respectively.
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at