Publications
Publications in peer reviewed journals
Microbial temperature sensitivity and biomass change explain soil carbon loss with warming.
2018 - Nat Clim Chang, 10: 885-889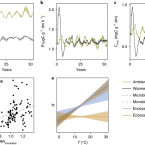
Abstract:
Soil microorganisms control carbon losses from soils to the atmosphere1-3, yet their responses to climate warming are often short-lived and unpredictable4-7. Two mechanisms, microbial acclimation and substrate depletion, have been proposed to explain temporary warming effects on soil microbial activity8-10. However, empirical support for either mechanism is unconvincing. Here we used geothermal temperature gradients (> 50 years of field warming)11 and a short-term experiment to show that microbial activity (gross rates of growth, turnover, respiration and carbon uptake) is intrinsically temperature sensitive and does not acclimate to warming (+ 6 ºC) over weeks or decades. Permanently accelerated microbial activity caused carbon loss from soil. However, soil carbon loss was temporary because substrate depletion reduced microbial biomass and constrained the influence of microbes over the ecosystem. A microbial biogeochemical model12-14 showed that these observations are reproducible through a modest, but permanent, acceleration in microbial physiology. These findings reveal a mechanism by which intrinsic microbial temperature sensitivity and substrate depletion together dictate warming effects on soil carbon loss their control over microbial biomass. We thus provide a framework for interpreting the links between temperature, microbial activity and soil carbon loss on timescales relevant to Earth's climate system.
Biodegradation of synthetic polymers in soils: Tracking carbon into CO2 and microbial biomass
2018 - Science Advances, 4: eaas9024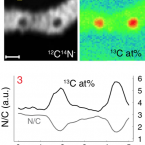
Abstract:
Plastic materials are widely used in agricultural applications to achieve food security for the growing world population. The use of biodegradable instead of nonbiodegradable polymers in single-use agricultural applications, including plastic mulching, promises to reduce plastic accumulation in the environment. We present a novel approach that allows tracking of carbon from biodegradable polymers into CO2 and microbial biomass. The approach is based on 13C-labeled polymers and on isotope-specific analytical methods, including nanoscale secondary ion mass spectrometry (NanoSIMS). Our results unequivocally demonstrate the biodegradability of poly(butylene adipate-co-terephthalate) (PBAT), an important polyester used in agriculture, in soil. Carbon from each monomer unit of PBAT was used by soil microorganisms, including filamentous fungi, to gain energy and to form biomass. This work advances both our conceptual understanding of polymer biodegradation and the methodological capabilities to assess this process in natural and engineered environments.
Recognizing Patterns: Spatial Analysis of Observed Microbial Colonization on Root Surfaces
2018 - Front Environ Sci, 6: 1-12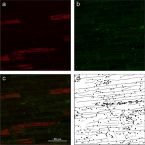
Abstract:
Root surfaces are major sites of interactions between plants and associated microorganisms. Here, plants and microbes communicate via signaling molecules, compete for nutrients, and release substrates that may have beneficial or harmful effects on each other. Whilst the body of knowledge on the abundance and diversity of microbial communities at root-soil interfaces is now substantial, information on their spatial distribution at the microscale is still scarce.
In this study, a standardized method for recognizing and analyzing microbial cell distributions on root surfaces is presented. Fluorescence microscopy was combined with automated image analysis and spatial statistics to explore the distribution of bacterial colonization patterns on rhizoplanes of rice roots. To test and evaluate the presented approach, a gnotobiotic experiment was performed using a potential nitrogen-fixing bacterial strain in combination with roots of wetland rice.
The automated analysis procedure resulted in reliable spatial data of bacterial cells colonizing the rhizoplane. Among all replicate roots, the analysis revealed an increasing density of bacterial cells from the root tip to the region of root cell maturation. Moreover, bacterial cells showed significant spatial clustering and tended to be located around plant root cell walls. The quantitative data suggest that the structure of the root surface plays a major role in bacterial colonization patterns.
Possible adaptations of the presented approach for future studies are discussed along with potential pitfalls such as inaccurate imaging. Our results demonstrate that standardized recognition and statistical evaluation of microbial colonization on root surfaces holds the potential to increase our understanding of microbial associations with roots and of the underlying ecological interactions.Evaluation of primers targeting the diazotroph functional gene and development of NifMAP – a bioinformatics pipeline for analyzing nifH amplicon data
2018 - Front Microbiol, 9: 1-15
Abstract:
Diazotrophic microorganisms introduce biologically available nitrogen (N) to the global N cycle through the activity of the nitrogenase enzyme. The genetically conserved dinitrogenase reductase (nifH) gene is phylogenetically distributed across four clusters (I-IV) and is widely used as a marker gene for N2 fixation, permitting investigators to study the genetic diversity of diazotrophs in nature and target potential participants in N2 fixation. To date there have been limited, standardized pipelines for the nifH functional gene, which is in stark contrast to the rRNA gene. Here we present a bioinformatics pipeline for processing nifH amplicon datasets – NifMAP (“NifH MiSeq Illumina amplicon Analysis Pipeline”), which as a novel aspect uses Hidden-Markov models to filter out homologous genes to nifH. By using this pipeline, we evaluated the broadly inclusive primer pairs (Ueda19F-R6, IGK3-DVV, F2-R6) that target the nifH gene. To evaluate any systematic biases, the nifH gene was amplified with the aforementioned primer pairs in a diverse collection of environmental samples (soils, rhizosphere and roots samples, biological soil crusts and estuarine samples), in addition to a nifH mock community consisting of six phylogenetically diverse members. We noted that all primer pairs co-amplified nifH homologs to varying degrees; up to 90% of the amplicons were nifH homologs with IGK3-DVV in some samples (rhizosphere and roots from tall oat-grass). In regards to specificity, we observed some degree of bias across the primer pairs. For example, primer pair F2-R6 discriminated against cyanobacteria (amongst others), yet captured many sequences from subclusters IIIE and IIIL-N. These aforementioned subclusters were largely missing by the primer pair IGK3-DVV, which also tended to discriminate against Alphaproteobacteria, but amplified sequences within clusters IIIC (affiliated with Clostridia) and clusters IVB and IVC. Primer pair Ueda19F-R6 exhibited the least bias and successfully captured diazotrophs in cluster I and subclusters IIIE, IIIL, IIIM and IIIN, but discriminated against Firmicutes and subcluster IIIC. Taken together, our newly established bioinformatics pipeline, NifMAP, along with our systematic evaluations of nifH primer pairs permit more robust, high-throughput investigations of diazotrophs in diverse environments.
Transmission of fungal partners to incipient Cecropia-tree ant colonies
2018 - PLoS One, 13: e0192207
Abstract:
Ascomycete fungi in the nests of ants inhabiting plants (= myrmecophytes) are very often cultivated by the ants in small patches and used as food source. Where these fungi come from is not known yet. Two scenarios of fungus recruitment are possible: (1) random infection through spores or hyphal fragments from the environment, or (2) transmission from mother to daughter colonies by the foundress queen. It is also not known at which stage of the colony life cycle fungiculture is initiated, and whether the- symbiont fungi serve as food for the ant queen. To clarify these questions, we investigated four Azteca ant species inhabiting three different Cecropia species (C. insignis, C. obtusifolia, and C. peltata). We analysed an rRNA gene fragment from 52 fungal patches produced by founding queens and compared them with those from established Azteca colonies (n = 54). The infrabuccal pockets of winged queens were dissected to investigate whether young queens carry fungi from their mother colony. Additionally, 15N labelling experiments were done to verify whether the queen feeds on the patches until she is nourished by her first worker offspring. We infer from the results that the fungi cultivated in hollow plant structures are transferred from the parental colony of the young queen. First, fungal genotypes/OTU diversity was not significantly different between foundress queen patches and established colonies, and second, hyphal parts were discovered in the infrabuccal pockets of female alates. We could show that fungiculture already starts before queens lay their eggs, and that the queens do not feed on fungal patch material but feed it to the larvae. Our findings suggest that fungiculture may be crucial for successful colony founding of arboreal ants in the tropics.
Peatland Acidobacteria with a dissimilatory sulfur metabolism
2018 - ISME J, 12: 1729-1742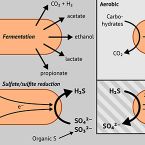
Abstract:
Sulfur-cycling microorganisms impact organic matter decomposition in wetlands and consequently greenhouse gas emissions from these globally relevant environments. However, their identities and physiological properties are largely unknown. By applying a functional metagenomics approach to an acidic peatland, we recovered draft genomes of seven novel Acidobacteria species with the potential for dissimilatory sulfite (dsrAB, dsrC, dsrD, dsrN, dsrT, dsrMKJOP) or sulfate respiration (sat, aprBA, qmoABC plus dsr genes). Surprisingly, the genomes also encoded DsrL, which so far was only found in sulfur-oxidizing microorganisms. Metatranscriptome analysis demonstrated expression of acidobacterial sulfur-metabolism genes in native peat soil and their upregulation in diverse anoxic microcosms. This indicated an active sulfate respiration pathway, which, however, might also operate in reverse for dissimilatory sulfur oxidation or disproportionation as proposed for the sulfur-oxidizing Desulfurivibrio alkaliphilus. Acidobacteria that only harbored genes for sulfite reduction additionally encoded enzymes that liberate sulfite from organosulfonates, which suggested organic sulfur compounds as complementary energy sources. Further metabolic potentials included polysaccharide hydrolysis and sugar utilization, aerobic respiration, several fermentative capabilities, and hydrogen oxidation. Our findings extend both, the known physiological and genetic properties of Acidobacteria and the known taxonomic diversity of microorganisms with a DsrAB-based sulfur metabolism, and highlight new fundamental niches for facultative anaerobic Acidobacteria in wetlands based on exploitation of inorganic and organic sulfur molecules for energy conservation.
Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments
2018 - Environ Microbiol, 20: 1041-1063
Abstract:
Members of the phylum Acidobacteria are abundant and ubiquitous across soils. We performed the largest (to date) comparative genome analysis spanning subdivisions 1, 3, 4, 6, 8, and 23 (n=24) with the goal to identify features to help explain their prevalence in soils and understand their ecophysiology. In contrast to earlier studies, our analysis revealed that bacteriophage integration events along with transposable and mobile elements influenced the structure and plasticity of these genomes. Low- and high-affinity respiratory oxygen reductases were detected in multiple genomes, suggesting the capacity for growing across different oxygen gradients. Amongst many genomes, the capacity to use a diverse collection of carbohydrates, as well as inorganic and organic N sources (such as extracellular peptidases), were detected – both advantageous traits in environments with fluctuating nutrient environments. We also identified multiple soil acidobacteria with the potential to scavenge atmospheric concentrations of H2, now encompassing mesophilic soil strains within the subdivision 1 and 3, in addition to a previously identified thermophilic strain in subdivision 4. This large-scale acidobacteria genome analysis reveals traits that provide genomic, physiological and metabolic versatility, presumably allowing flexibility and versatility in the challenging and fluctuating soil environment.
Application of stable-isotope labelling techniques for the detection of active diazotrophs
2018 - Environmental Microbiology, 20: 44-61
Abstract:
Investigating active participants in the fixation of dinitrogen gas is vital as N is often a limiting factor for primary production. Biological nitrogen fixation (BNF) is performed by a diverse guild of bacteria and archaea (diazotrophs), which can be free-living or symbionts. Free-living diazotrophs are widely distributed in the environment, yet our knowledge about their identity and ecophysiology is still limited. A major challenge in investigating this guild is inferring activity from genetic data as this process is highly regulated. To address this challenge, we evaluated and improved several 15N-based methods for detecting N2 fixation activity (with a focus on soil samples) and studying active diazotrophs. We compared the acetylene reduction assay and the 15N2 tracer method and demonstrated that the latter is more sensitive in samples with low activity. Additionally, tracing 15N into microbial RNA provides much higher sensitivity compared to bulk soil analysis. Active soil diazotrophs were identified with a 15N-RNA-SIP approach optimized for environmental samples and benchmarked to 15N-DNA-SIP. Lastly, we investigated the feasibility of using SIP-Raman microspectroscopy for detecting 15N-labelled cells. Taken together, these tools allow identifying and investigating active free-living diazotrophs in a highly sensitive manner in diverse environments, from bulk to the single-cell level.
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at