Publications
Publications in peer reviewed journals
Comparison of genovars and Chlamydia trachomatis infection loads in ocular samples from children in two distinct cohorts in Sudan and Morocco.
2021 - PLoS Negl Trop Dis, 8: e0009655
Abstract:
Trachoma is a blinding disease caused by repeated conjunctival infection with different Chlamydia trachomatis (Ct) genovars. Ct B genovars have been associated with more severe trachoma symptoms. Here, we investigated associations between Ct genovars and bacterial loads in ocular samples from two distinct geographical locations in Africa, which are currently unclear. We tested ocular swabs from 77 Moroccan children (28 with trachomatous inflammation-follicular (TF) and 49 healthy controls), and 96 Sudanese children (54 with TF and 42 healthy controls) with a Ct-specific real-time polymerase chain reaction (PCR) assay. To estimate bacterial loads, Ct-positive samples were further processed by multiplex real-time qPCR to amplify the chromosomal outer membrane complex B and plasmid open reading frame 2 of Ct. Genotyping was performed by PCR-based amplification of the outer membrane protein A gene (~1120 base pairs) of Ct and Sanger sequencing. Ct-positivities among the Moroccan and Sudanese patient groups were 60·7% and 31·5%, respectively. Significantly more Sudanese patients than Moroccan patients were genovar A-positive. In contrast, B genovars were significantly more prevalent in Moroccan patients than in Sudanese patients. Significantly higher Ct loads were found in samples positive for B genovars (598·596) than A genovar (51·005). Geographical differences contributed to the distributions of different ocular Ct genovars. B genovars may induce a higher bacterial load than A genovars in trachoma patients. Our findings emphasize the importance of conducting broader studies to elucidate if the noted difference in multiplication abilities are genovar and/or endemicity level dependent.
Pangenomics reveals alternative environmental lifestyles among chlamydiae
2021 - Nature Commun, 12: 4021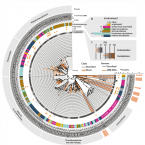
Abstract:
Chlamydiae are highly successful strictly intracellular bacteria associated with diverse eukaryotic hosts. Here we analysed metagenome-assembled genomes of the “Genomes from Earth’s Microbiomes” initiative from diverse environmental samples, which almost double the known phylogenetic diversity of the phylum and facilitate a highly resolved view at the chlamydial pangenome. Chlamydiae are defined by a relatively large core genome indicative of an intracellular lifestyle, and a highly dynamic accessory genome of environmental lineages. We observe chlamydial lineages that encode enzymes of the reductive tricarboxylic acid cycle and for light-driven ATP synthesis. We show a widespread potential for anaerobic energy generation through pyruvate fermentation or the arginine deiminase pathway, and we add lineages capable of molecular hydrogen production. Genome-informed analysis of environmental distribution revealed lineage-specific niches and a high abundance of chlamydiae in some habitats. Together, our data provide an extended perspective of the variability of chlamydial biology and the ecology of this phylum of intracellular microbes.
Coevolving plasmids drive gene flow and genome plasticity in host-associated intracellular bacteria
2021 - Curr Biol, 2: 346-357.e3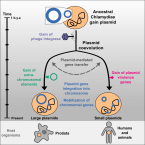
Abstract:
Plasmids are important in microbial evolution and adaptation to new environments. Yet, carrying a plasmid can be costly, and long-term association of plasmids with their hosts is poorly understood. Here, we provide evidence that the Chlamydiae, a phylum of strictly host-associated intracellular bacteria, have coevolved with their plasmids since their last common ancestor. Current chlamydial plasmids are amalgamations of at least one ancestral plasmid and a bacteriophage. We show that the majority of plasmid genes are also found on chromosomes of extant chlamydiae. The most conserved plasmid gene families are predominantly vertically inherited, while accessory plasmid gene families show significantly increased mobility. We reconstructed the evolutionary history of plasmid gene content of an entire bacterial phylum over a period of around one billion years. Frequent horizontal gene transfer and chromosomal integration events illustrate the pronounced impact of coevolution with these extrachromosomal elements on bacterial genome dynamics in host-dependent microbes.
Book chapters and other publications
A genomic catalog of Earth's microbiomes
2021 - Nat Biotechnol, 39: 499-509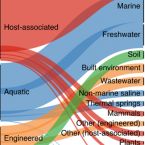
Abstract:
The reconstruction of bacterial and archaeal genomes from shotgun metagenomes has enabled insights into the ecology and evolution of environmental and host-associated microbiomes. Here we applied this approach to >10,000 metagenomes collected from diverse habitats covering all of Earth's continents and oceans, including metagenomes from human and animal hosts, engineered environments, and natural and agricultural soils, to capture extant microbial, metabolic and functional potential. This comprehensive catalog includes 52,515 metagenome-assembled genomes representing 12,556 novel candidate species-level operational taxonomic units spanning 135 phyla. The catalog expands the known phylogenetic diversity of bacteria and archaea by 44% and is broadly available for streamlined comparative analyses, interactive exploration, metabolic modeling and bulk download. We demonstrate the utility of this collection for understanding secondary-metabolite biosynthetic potential and for resolving thousands of new host linkages to uncultivated viruses. This resource underscores the value of genome-centric approaches for revealing genomic properties of uncultivated microorganisms that affect ecosystem processes.
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at