Publications
Publications in peer reviewed journals
Global diversity and inferred ecophysiology of microorganisms with the potential for dissimilatory sulfate/sulfite reduction.
2023 - FEMS Microbiol Rev, fuad058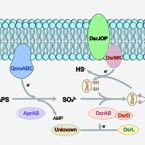
Abstract:
Sulfate/sulfite-reducing microorganisms (SRM) are ubiquitous in nature, driving the global sulfur cycle. A hallmark of SRM is the dissimilatory sulfite reductase encoded by the genes dsrAB. Based on analysis of 950 mainly metagenome-derived dsrAB-carrying genomes, we redefine the global diversity of microorganisms with the potential for dissimilatory sulfate/sulfite reduction and uncover genetic repertoires that challenge earlier generalizations regarding their mode of energy metabolism. We show: (i) 19 out of 23 bacterial and 2 out of 4 archaeal phyla harbor uncharacterized SRM, (ii) four phyla including the Desulfobacterota harbor microorganisms with the genetic potential to switch between sulfate/sulfite reduction and sulfur oxidation, and (iii) the combination as well as presence/absence of different dsrAB-types, dsrL-types and dsrD provides guidance on the inferred direction of dissimilatory sulfur metabolism. We further provide an updated dsrAB database including >60% taxonomically resolved, uncultured family-level lineages and recommendations on existing dsrAB-targeted primers for environmental surveys. Our work summarizes insights into the inferred ecophysiology of newly discovered SRM, puts SRM diversity into context of the major recent changes in bacterial and archaeal taxonomy, and provides an up-to-date framework to study SRM in a global context.
Ecophysiology and interactions of a taurine-respiring bacterium in the mouse gut.
2023 - Nat Commun, 1: 5533
Abstract:
Taurine-respiring gut bacteria produce HS with ambivalent impact on host health. We report the isolation and ecophysiological characterization of a taurine-respiring mouse gut bacterium. Taurinivorans muris strain LT0009 represents a new widespread species that differs from the human gut sulfidogen Bilophila wadsworthia in its sulfur metabolism pathways and host distribution. T. muris specializes in taurine respiration in vivo, seemingly unaffected by mouse diet and genotype, but is dependent on other bacteria for release of taurine from bile acids. Colonization of T. muris in gnotobiotic mice increased deconjugation of taurine-conjugated bile acids and transcriptional activity of a sulfur metabolism gene-encoding prophage in other commensals, and slightly decreased the abundance of Salmonella enterica, which showed reduced expression of galactonate catabolism genes. Re-analysis of metagenome data from a previous study further suggested that T. muris can contribute to protection against pathogens by the commensal mouse gut microbiota. Together, we show the realized physiological niche of a key murine gut sulfidogen and its interactions with selected gut microbiota members.
Secondary Metabolite Production Potential in a Microbiome of the Freshwater Sponge Spongilla lacustris
2023 - Microbiol Spectr, e0435322
Abstract:
Marine and freshwater sponges harbor diverse communities of bacteria with vast potential to produce secondary metabolites that may play an important role in protecting the host from predators and infections. In this work, we initially used cultivation and metagenomics to investigate the microbial community of the freshwater sponge Spongilla lacustris collected in an Austrian lake. Representatives of 41 bacterial genera were isolated from the sponge sample and classified according to their 16S rRNA gene sequences. The genomes of 33 representative isolates and the 20 recovered metagenome-assembled genomes (MAGs) contained in total 306 secondary metabolite biosynthesis gene clusters (BGCs). Comparative 16S rRNA gene and genome analyses showed very little taxon overlap between the recovered isolates and the sponge community as revealed by cultivation-independent methods. Both culture-independent and -dependent analyses suggested high biosynthetic potential of the S. lacustris microbiome, which was confirmed experimentally even at the subspecies level for two isolates. To our knowledge, this is the most thorough description of the secondary metabolite production potential of a freshwater sponge microbiome to date. A large body of research is dedicated to marine sponges, filter-feeding animals harboring rich bacterial microbiomes believed to play an important role in protecting the host from predators and infections. Freshwater sponges have received so far much less attention with respect to their microbiomes, members of which may produce bioactive secondary metabolites with potential to be developed into drugs to treat a variety of diseases. In this work, we investigated the potential of bacteria associated with the freshwater sponge to biosynthesize diverse secondary metabolites. Using culture-dependent and -independent methods, we discovered over 300 biosynthetic gene clusters in sponge-associated bacteria and proved production of several compounds by selected isolates using genome mining. Our results illustrate the importance of a complex approach when dealing with microbiomes of multicellular organisms that may contain producers of medically important secondary metabolites.
Sulfur and methane oxidation by a single microorganism.
2022 - Proc Natl Acad Sci U S A, 32: e2114799119
Abstract:
Natural and anthropogenic wetlands are major sources of the atmospheric greenhouse gas methane. Methane emissions from wetlands are mitigated by methanotrophic bacteria at the oxic-anoxic interface, a zone of intense redox cycling of carbon, sulfur, and nitrogen compounds. Here, we report on the isolation of an aerobic methanotrophic bacterium, '' strain HY1, which possesses metabolic capabilities never before found in any methanotroph. Most notably, strain HY1 is the first bacterium shown to aerobically oxidize both methane and reduced sulfur compounds for growth. Genomic and proteomic analyses showed that soluble methane monooxygenase and XoxF-type alcohol dehydrogenases are responsible for methane and methanol oxidation, respectively. Various pathways for respiratory sulfur oxidation were present, including the Sox-rDsr pathway and the SI system. Strain HY1 employed the Calvin-Benson-Bassham cycle for CO fixation during chemolithoautotrophic growth on reduced sulfur compounds. Proteomic and microrespirometry analyses showed that the metabolic pathways for methane and thiosulfate oxidation were induced in the presence of the respective substrates. Methane and thiosulfate could therefore be independently or simultaneously oxidized. The discovery of this versatile bacterium demonstrates that methanotrophy and thiotrophy are compatible in a single microorganism and underpins the intimate interactions of methane and sulfur cycles in oxic-anoxic interface environments.
Genomic insights into diverse bacterial taxa that degrade extracellular DNA in marine sediments
2021 - Nat Microbiol, 6: 885–898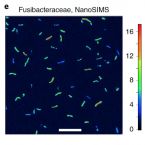
Abstract:
Extracellular DNA is a major macromolecule in global element cycles, and is a particularly crucial phosphorus, nitrogen and carbon source for microorganisms in the seafloor. Nevertheless, the identities, ecophysiology and genetic features of DNA-foraging microorganisms in marine sediments are largely unknown. Here, we combined microcosm experiments, DNA stable isotope probing (SIP), single-cell SIP using nano-scale secondary isotope mass spectrometry (NanoSIMS) and genome-centric metagenomics to study microbial catabolism of DNA and its subcomponents in marine sediments. 13C-DNA added to sediment microcosms was largely degraded within 10 d and mineralized to 13CO2. SIP probing of DNA revealed diverse ‘Candidatus Izemoplasma’, Lutibacter, Shewanella and Fusibacteraceae incorporated DNA-derived 13C-carbon. NanoSIMS confirmed incorporation of 13C into individual bacterial cells of Fusibacteraceae sorted from microcosms. Genomes of the 13C-labelled taxa all encoded enzymatic repertoires for catabolism of DNA or subcomponents of DNA. Comparative genomics indicated that diverse ‘Candidatus Izemoplasmatales’ (former Tenericutes) are exceptional because they encode multiple (up to five) predicted extracellular nucleases and are probably specialized DNA-degraders. Analyses of additional sediment metagenomes revealed extracellular nuclease genes are prevalent among Bacteroidota at diverse sites. Together, our results reveal the identities and functional properties of microorganisms that may contribute to the key ecosystem function of degrading and recycling DNA in the seabed.
Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling.
2021 - ISME J, 15: 3159–3180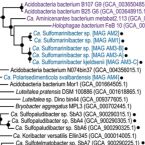
Abstract:
Acidobacteriota are widespread and often abundant in marine sediments, yet their metabolic and ecological properties are poorly understood. Here, we examined metabolisms and distributions of Acidobacteriota in marine sediments of Svalbard by functional predictions from metagenome-assembled genomes (MAGs), amplicon sequencing of 16S rRNA and dissimilatory sulfite reductase (dsrB) genes and transcripts, and gene expression analyses of tetrathionate-amended microcosms. Acidobacteriota were the second most abundant dsrB-harboring (averaging 13%) phylum after Desulfobacterota in Svalbard sediments, and represented 4% of dsrB transcripts on average. Meta-analysis of dsrAB datasets also showed Acidobacteriota dsrAB sequences are prominent in marine sediments worldwide, averaging 15% of all sequences analysed, and represent most of the previously unclassified dsrAB in marine sediments. We propose two new Acidobacteriota genera, Candidatus Sulfomarinibacter (class Thermoanaerobaculia, "subdivision 23") and Ca. Polarisedimenticola ("subdivision 22"), with distinct genetic properties that may explain their distributions in biogeochemically distinct sediments. Ca. Sulfomarinibacter encode flexible respiratory routes, with potential for oxygen, nitrous oxide, metal-oxide, tetrathionate, sulfur and sulfite/sulfate respiration, and possibly sulfur disproportionation. Potential nutrients and energy include cellulose, proteins, cyanophycin, hydrogen, and acetate. A Ca. Polarisedimenticola MAG encodes various enzymes to degrade proteins, and to reduce oxygen, nitrate, sulfur/polysulfide and metal-oxides. 16S rRNA gene and transcript profiling of Svalbard sediments showed Ca. Sulfomarinibacter members were relatively abundant and transcriptionally active in sulfidic fjord sediments, while Ca. Polarisedimenticola members were more relatively abundant in metal-rich fjord sediments. Overall, we reveal various physiological features of uncultured marine Acidobacteriota that indicate fundamental roles in seafloor biogeochemical cycling.
An economical and flexible dual barcoding, two-step PCR approach for highly multiplexed amplicon sequencing
2021 - Front Microbiol, 12: 669776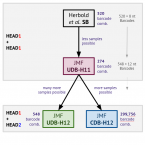
Abstract:
In microbiome research, phylogenetic and functional marker gene amplicon sequencing is the most commonly-used community profiling approach. Consequently, a plethora of protocols for the preparation and multiplexing of samples for amplicon sequencing have been developed. Here, we present two economical high-throughput gene amplification and sequencing workflows that are implemented as standard operating procedures at the Joint Microbiome Facility of the Medical University of Vienna and the University of Vienna. These workflows are based on a previously-published two-step PCR approach, but have been updated to either increase the accuracy of results, or alternatively to achieve orders of magnitude higher numbers of samples to be multiplexed in a single sequencing run. The high-accuracy workflow relies on unique dual sample barcoding. It allows the same level of sample multiplexing as the previously-published two-step PCR approach, but effectively eliminates residual read missasignments between samples (crosstalk) which are inherent to single barcoding approaches. The high-multiplexing workflow is based on combinatorial dual sample barcoding, which theoretically allows for multiplexing up to 299,756 amplicon libraries of the same target gene in a single massively-parallelized amplicon sequencing run. Both workflows presented here are highly economical, easy to implement, and can, without significant modifications or cost, be applied to any target gene of interest.
Sulfoquinovose is a select nutrient of prominent bacteria and a source of hydrogen sulfide in the human gut.
2021 - ISME J, 15: 2779–2791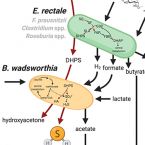
Abstract:
Responses of the microbiota to diet are highly personalized but mechanistically not well understood because many metabolic capabilities and interactions of human gut microorganisms are unknown. Here we show that sulfoquinovose (SQ), a sulfonated monosaccharide omnipresent in green vegetables, is a selective yet relevant substrate for few but ubiquitous bacteria in the human gut. In human feces and in defined co-culture, Eubacterium rectale and Bilophila wadsworthia used recently identified pathways to cooperatively catabolize SQ with 2,3-dihydroxypropane-1-sulfonate as a transient intermediate to hydrogen sulfide (HS), a key intestinal metabolite with disparate effects on host health. SQ-degradation capability is encoded in almost half of E. rectale genomes but otherwise sparsely distributed among microbial species in the human intestine. However, re-analysis of fecal metatranscriptome datasets of four human cohorts showed that SQ degradation (mostly from E. rectale and Faecalibacterium prausnitzii) and HS production (mostly from B. wadsworthia) pathways were expressed abundantly across various health states, demonstrating that these microbial functions are core attributes of the human gut. The discovery of green-diet-derived SQ as an exclusive microbial nutrient and an additional source of HS in the human gut highlights the role of individual dietary compounds and organosulfur metabolism on microbial activity and has implications for precision editing of the gut microbiota by dietary and prebiotic interventions.
Anaerobic bacterial degradation of protein and lipid macromolecules in subarctic marine sediment
2021 - ISME J, 15: 833-847
Abstract:
Microorganisms in marine sediments play major roles in marine biogeochemical cycles by mineralizing substantial quantities of organic matter from decaying cells. Proteins and lipids are abundant components of necromass, yet the taxonomic identities of microorganisms that actively degrade them remain poorly resolved. Here, we revealed identities, trophic interactions and genomic features of bacteria that degraded 13C-labelled proteins and lipids in cold anoxic microcosms containing sulfidic subarctic marine sediment. Supplemented proteins and lipids were rapidly fermented to various volatile fatty acids within five days. DNA-stable isotope probing (SIP) suggested Psychrilyobacter atlanticus was an important primary degrader of proteins, and Psychromonas members were important primary degraders of both proteins and lipids. Closely related Psychromonas populations, as represented by distinct 16S rRNA gene variants, differentially utilized either proteins or lipids. DNA-SIP also showed 13C-labeling of various Deltaproteobacteria within ten days, indicating trophic transfer of carbon to putative sulfate-reducers. Metagenome-assembled genomes revealed the primary hydrolyzers encoded secreted peptidases or lipases, and enzymes for catabolism of protein or lipid degradation products. Psychromonas species are prevalent in diverse marine sediments, suggesting they are important players in organic carbon processing in situ. Together, this study provides new insights into the identities, functions and genomes of bacteria that actively degrade abundant necromass macromolecules in the seafloor.
Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities
2020 - Int J Syst Evol Microbiol, 11: 5972-6016
Abstract:
The class comprises an ecologically and metabolically diverse group of bacteria best known for dissimilatory sulphate reduction and predatory behaviour. Although this lineage is the fourth described class of the phylum , it rarely affiliates with other proteobacterial classes and is frequently not recovered as a monophyletic unit in phylogenetic analyses. Indeed, one branch of the class encompassing like predators was recently reclassified into a separate proteobacterial class, the . Here we systematically explore the phylogeny of taxa currently assigned to these classes using 120 conserved single-copy marker genes as well as rRNA genes. The overwhelming majority of markers reject the inclusion of the classes and in the phylum . Instead, the great majority of currently recognized members of the class are better classified into four novel phylum-level lineages. We propose the names phyl. nov. and phyl. nov. for two of these phyla, based on the oldest validly published names in each lineage, and retain the placeholder name SAR324 for the third phylum pending formal description of type material. Members of the class represent a separate phylum for which we propose the name phyl. nov. based on priority in the literature and general recognition of the genus phyl. nov. includes the taxa previously classified in the phylum , and these reclassifications imply that the ability of sulphate reduction was vertically inherited in the rather than laterally acquired as previously inferred. Our analysis also indicates the independent acquisition of predatory behaviour in the phyla and , which is consistent with their distinct modes of action. This work represents a stable reclassification of one of the most taxonomically challenging areas of the bacterial tree and provides a robust framework for future ecological and systematic studies.
Environmental and intestinal phylum Firmicutes bacteria metabolize the plant sugar sulfoquinovose via a 6-deoxy-6-sulfofructose transaldolase pathway
2020 - iScience, 23: 101510
Abstract:
Bacterial degradation of the sugar sulfoquinovose (SQ, 6-deoxy-6-sulfoglucose) produced by plants, algae and cyanobacteria, is an important component of the biogeochemical carbon and sulfur cycles. Here, we reveal a third biochemical pathway for primary SQ degradation in an aerobic Bacillus aryabhattaistrain. An isomerase converts SQ to 6-deoxy-6-sulfofructose (SF). A novel transaldolase enzyme cleaves the SF to 3-sulfolactaldehyde (SLA), while the non-sulfonated C3-(glycerone)-moiety is transferred to an acceptor molecule, glyceraldehyde phosphate (GAP), yielding fructose-6-phosphate (F6P). Intestinal anaerobic bacteria such as Enterococcus gilvus, Clostridium symbiosum and Eubacterium rectale strains also express transaldolase-pathway gene clusters during fermentative growth with SQ. The now three known biochemical strategies for SQ catabolism reflect adaptations to the aerobic or anaerobic life-style of the different bacteria. The occurrence of these pathways in intestinal (family) Enterobacteriaceae and (phylum) Firmicutes strains further highlights a potential importance of metabolism of green-diet SQ by gut microbial communities to, ultimately, hydrogen sulfide.
Woeseiales transcriptional response to shallow burial in Arctic fjord surface sediment
2020 - PLoS One, 15: e0234839
Abstract:
Distinct lineages of Gammaproteobacteria clade Woeseiales are globally distributed in marine sediments, based on metagenomic and 16S rRNA gene analysis. Yet little is known about why they are dominant or their ecological role in Arctic fjord sediments, where glacial retreat is rapidly imposing change. This study combined 16S rRNA gene analysis, metagenome-assembled genomes (MAGs), and genome-resolved metatranscriptomics uncovered the in situ abundance and transcriptional activity of Woeseiales with burial in four shallow sediment sites of Kongsfjorden and Van Keulenfjorden of Svalbard (79°N). We present five novel Woeseiales MAGs and show transcriptional evidence for metabolic plasticity during burial, including sulfur oxidation with reverse dissimilatory sulfite reductase (dsrAB) down to 4 cm depth and nitrite reduction down to 6 cm depth. A single stress protein, spore protein SP21 (hspA), had a tenfold higher mRNA abundance than any other transcript, and was a hundredfold higher on average than other transcripts. At three out of the four sites, SP21 transcript abundance increased with depth, while total mRNA abundance and richness decreased, indicating a shift in investment from metabolism and other cellular processes to build-up of spore protein SP21. The SP21 gene in MAGs was often flanked by genes involved in membrane-associated stress response. The ability of Woeseiales to shift from sulfur oxidation to nitrite reduction with burial into marine sediments with decreasing access to overlying oxic bottom waters, as well as enter into a dormant state dominated by SP21, may account for its ubiquity and high abundance in marine sediments worldwide, including those of the rapidly shifting Arctic.
Microbiome definition re-visited: old concepts and new challenges.
2020 - Microbiome, 1: 103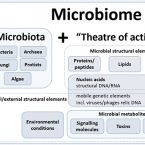
Abstract:
The field of microbiome research has evolved rapidly over the past few decades and has become a topic of great scientific and public interest. As a result of this rapid growth in interest covering different fields, we are lacking a clear commonly agreed definition of the term "microbiome." Moreover, a consensus on best practices in microbiome research is missing. Recently, a panel of international experts discussed the current gaps in the frame of the European-funded MicrobiomeSupport project. The meeting brought together about 40 leaders from diverse microbiome areas, while more than a hundred experts from all over the world took part in an online survey accompanying the workshop. This article excerpts the outcomes of the workshop and the corresponding online survey embedded in a short historical introduction and future outlook. We propose a definition of microbiome based on the compact, clear, and comprehensive description of the term provided by Whipps et al. in 1988, amended with a set of novel recommendations considering the latest technological developments and research findings. We clearly separate the terms microbiome and microbiota and provide a comprehensive discussion considering the composition of microbiota, the heterogeneity and dynamics of microbiomes in time and space, the stability and resilience of microbial networks, the definition of core microbiomes, and functionally relevant keystone species as well as co-evolutionary principles of microbe-host and inter-species interactions within the microbiome. These broad definitions together with the suggested unifying concepts will help to improve standardization of microbiome studies in the future, and could be the starting point for an integrated assessment of data resulting in a more rapid transfer of knowledge from basic science into practice. Furthermore, microbiome standards are important for solving new challenges associated with anthropogenic-driven changes in the field of planetary health, for which the understanding of microbiomes might play a key role. Video Abstract.
Hair eruption initiates and commensal skin microbiota aggravate adverse events of anti-EGFR therapy
2019 - Sci Transl Med, 11: eaax2693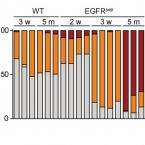
Abstract:
Epidermal growth factor receptor (EGFR)–targeted anticancer therapy induces stigmatizing skin toxicities affecting patients’ quality of life and therapy adherence. The lack of mechanistic details underlying these adverse events hampers their management. We found that EGFR/ERK signaling is required in LRIG1-positive stem cells during de novo hair eruption to secure barrier integrity and prevent the invasion of commensal microbiota and inflammatory skin disease. EGFR-deficient epidermis is permissive for microbiota outgrowth and displays an atopic-like TH2-dominated signature. The opening of the follicular ostia during hair eruption allows invasion of commensal microbiota into the hair follicle, initiating an additional TH1 and TH17 response culminating in chronic folliculitis. Restoration of epidermal ERK signaling via prophylactic FGF7 treatment or transgenic SOS expression rescues the barrier defect in the absence of EGFR, highlighting a therapeutic anchor point. These data reveal that commensal skin microbiota provoke atopic-like inflammatory skin diseases by invading into the follicular opening of erupting hair.
Glacial runoff promotes deep burial of sulfur cycling-associated microorganisms in marine sediments
2019 - Front Microbiol, 10: 2558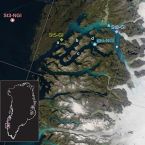
Abstract:
Marine fjords with active glacier outlets are hot spots for organic matter burial in the sediments and subsequent microbial mineralization. Here, we investigated controls on microbial community assembly in sub-arctic glacier-influenced (GI) and non-glacier-influenced (NGI) marine sediments in the Godthåbsfjord region, south-western Greenland. We used a correlative approach integrating 16S rRNA gene and dissimilatory sulfite reductase (dsrB) amplicon sequence data over six meters of depth with biogeochemistry, sulfur-cycling activities, and sediment ages. GI sediments were characterized by comparably high sedimentation rates and had ‘young’ sediment ages of <500 years even at 6 m sediment depth. In contrast, NGI stations reached ages of approximately 10,000 years at these depths. Sediment age-depth relationships, sulfate reduction rates, and C/N ratios were strongly correlated with differences in microbial community composition between GI and NGI sediments, indicating that age and diagenetic state were key drivers of microbial community assembly in subsurface sediments. Similar bacterial and archaeal communities were present in the surface sediments of all stations, whereas only in GI sediments were many surface taxa also abundant through the whole sediment core. The relative abundance of these taxa, including diverse Desulfobacteraceae members, correlated positively with sulfate reduction rates, indicating their active contributions to sulfur-cycling processes. In contrast, other surface community members, such as Desulfatiglans, Atribacteria and Chloroflexi, survived the slow sediment burial at NGI stations and dominated in the deepest sediment layers. These taxa are typical for the energy-limited marine deep biosphere and their relative abundances correlated positively with sediment age. In conclusion, our data suggests that high rates of sediment accumulation caused by glacier runoff and associated changes in biogeochemistry, promote persistence of sulfur-cycling activity and burial of a larger fraction of the surface microbial community into the deep subsurface.
A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome.
2019 - Nat Commun, 1: 4366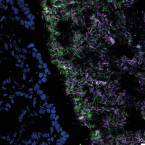
Abstract:
Compartmentalization of the gut microbiota is thought to be important to system function, but the extent of spatial organization in the gut ecosystem remains poorly understood. Here, we profile the murine colonic microbiota along longitudinal and lateral axes using laser capture microdissection. We found fine-scale spatial structuring of the microbiota marked by gradients in composition and diversity along the length of the colon. Privation of fiber reduces the diversity of the microbiota and disrupts longitudinal and lateral gradients in microbiota composition. Both mucus-adjacent and luminal communities are influenced by the absence of dietary fiber, with the loss of a characteristic distal colon microbiota and a reduction in the mucosa-adjacent community, concomitant with depletion of the mucus layer. These results indicate that diet has not only global but also local effects on the composition of the gut microbiota, which may affect function and resilience differently depending on location.
Draft Genome Sequence of Desulfosporosinus fructosivorans Strain 63.6F, Isolated from Marine Sediment in the Baltic Sea
2019 - Microbiology Resource Announcements, 8: e00427-1
Abstract:
Desulfosporosinus fructosivorans strain 63.6FT is a strictly anaerobic, spore-forming, sulfate-reducing bacterium isolated from marine sediment in the Baltic Sea. Here, we report the draft genome sequence of D. fructosivorans 63.6FT.
Diversity decoupled from sulfur isotope fractionation in a sulfate reducing microbial community
2019 - Geobiology, 17: 660-67
Abstract:
The extent of fractionation of sulfur isotopes by sulfate-reducing microbes is dictated by genomic and environmental factors. A greater understanding of species-specific fractionations may better inform interpretation of sulfur isotopes preserved in the rock record. To examine whether gene diversity influences net isotopic fractionation in situ, we assessed environmental chemistry, sulfate reduction rates, diversity of putative sulfur-metabolizing organisms by 16S rRNA and dissimilatory sulfite reductase (dsrB) gene amplicon sequencing, and net fractionation of sulfur isotopes along a sediment transect of a hypersaline Arctic spring. In situ sulfate reduction rates yielded minimum cell-specific sulfate reduction rates < 0.3 × 10-15 moles cell-1 day-1 . Neither 16S rRNA nor dsrB diversity indices correlated with relatively constant (38‰-45‰) net isotope fractionation (ε34 Ssulfide-sulfate ). Measured ε34 S values could be reproduced in a mechanistic fractionation model if 1%-2% of the microbial community (10%-60% of Deltaproteobacteria) were engaged in sulfate respiration, indicating heterogeneous respiratory activity within sulfate-reducing populations. This model indicated enzymatic kinetic diversity of Apr was more likely to correlate with sulfur fractionation than DsrB. We propose that, above a threshold Shannon diversity value of 0.8 for dsrB, the influence of the specific composition of the microbial community responsible for generating an isotope signal is overprinted by the control exerted by environmental variables on microbial physiology.
Draft Genome Sequence of Desulfosporosinus sp. Strain Sb-LF, Isolated from an acidic peatland in Germany
2019 - Microbiology Resource Announcements, 8: e00428-1
Abstract:
Desulfosporosinus sp. strain Sb-LF was isolated from an acidic peatland in Bavaria, Germany. Here, we report the draft genome sequence of the sulfate-reducing and lactate-utilizing strain Sb-LF.
Mucispirillum schaedleri antagonizes Salmonella virulence to protect mice against colitis
2019 - Cell Host Microbe, 25: 681-694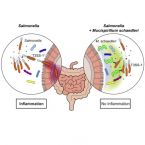
Abstract:
The microbiota and the gastrointestinal mucus layer play a pivotal role in protection against non-typhoidal Salmonellaenterica serovar Typhimurium (S. Tm) colitis. Here, we analyzed the course of Salmonella colitis in mice lacking a functional mucus layer in the gut. Unexpectedly, in contrast to mucus-proficient littermates, genetically deficient mice were protected against Salmonella-induced gut inflammation in the streptomycin colitis model. This correlated with microbiota alterations and enrichment of the bacterial phylum Deferribacteres. Using gnotobiotic mice associated with defined bacterial consortia, we causally linked Mucispirillum schaedleri, currently the sole known representative of Deferribacteres present in the mammalian microbiota, to host protection against S. Tm colitis. Inhibition by M. schaedleri involves interference with S. Tm invasion gene expression, partly by competing for anaerobic electron acceptors. In conclusion, this study establishes M. schaedleri, a core member of the murine gut microbiota, as a key antagonist of S. Tm virulence in the gut.
Historical factors associated with past environments influence the biogeography of thermophilic endospores in Arctic marine sediments
2019 - Front Microbiol, 10: 245
Abstract:
Selection by the local, contemporary environment plays a prominent role in shaping the biogeography of microbes. However, the importance of historical factors in microbial biogeography is more debatable. Historical factors include past ecological and evolutionary circumstances that may have influenced present-day microbial diversity, such as dispersal and past environmental conditions. Diverse thermophilic sulfate-reducing are present as dormant endospores in marine sediments worldwide where temperatures are too low to support their growth. Therefore, they are dispersed to here from elsewhere, presumably a hot, anoxic habitat. While dispersal through ocean currents must influence their distribution in cold marine sediments, it is not clear whether even earlier historical factors, related to the source habitat where these organisms were once active, also have an effect. We investigated whether these historical factors may have influenced the diversity and distribution of thermophilic endospores by comparing their diversity in 10 Arctic fjord surface sediments. Although community composition varied spatially, clear biogeographic patterns were only evident at a high level of taxonomic resolution (>97% sequence similarity of the 16S rRNA gene) achieved with oligotyping. In particular, the diversity and distribution of oligotypes differed for the two most prominent OTUs (defined using a standard 97% similarity cutoff). One OTU was dominated by a single ubiquitous oligotype, while the other OTU consisted of ten more spatially localized oligotypes that decreased in compositional similarity with geographic distance. These patterns are consistent with differences in historical factors that occurred when and where the taxa were once active, prior to sporulation. Further, the influence of history on biogeographic patterns was only revealed by analyzing microdiversity within OTUs, suggesting that populations within standard OTU-level groupings do not necessarily share a common ecological and evolutionary history.
Long-term transcriptional activity at zero growth by a cosmopolitan rare biosphere member
2019 - mBio, 10: e02189-18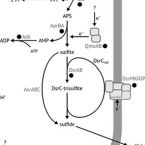
Abstract:
Microbial diversity in the environment is mainly concealed within the rare biosphere (all species with <0.1% relative abundance). While dormancy explains a low-abundance state very well, the mechanisms leading to rare but active microorganisms remain elusive. We used environmental systems biology to genomically and transcriptionally characterise Candidatus Desulfosporosinus infrequens, a low-abundance sulfate-reducing microorganism cosmopolitan to freshwater wetlands, where it contributes to cryptic sulfur cycling. We obtained its near-complete genome by metagenomics of acidic peat soil. In addition, we analyzed anoxic peat soil incubated under in situ-like conditions for 50 days by Desulfosporosinus-targeted qPCR and metatranscriptomics. The Desulfosporosinus population stayed at a constant low abundance under all incubation conditions, averaging 1.2 × 10⁶ 16S rRNA gene copies per cm³ soil. In contrast, transcriptional activity of Ca.D. infrequens increased at day 36 by 56- to 188-fold when minor amendments of acetate, propionate, lactate, or butyrate were provided with sulfate, as compared to the no-substrate-control. Overall transcriptional activity was driven by expression of genes encoding ribosomal proteins, energy metabolism and stress response but not by expression of genes encoding cell growth-associated processes. Since our results did not support growth of these highly active microorganisms in terms of biomass increase or cell division, they had to invest their sole energy for maintenance, most likely counterbalancing acidic pH conditions. This finding explains how a rare biosphere member can contribute to a biogeochemically relevant process while remaining in a zero growth state over a period of 50 days.
Stable-isotope probing of human and animal microbiome function
2018 - Trends Microbiol, 26: 999-1007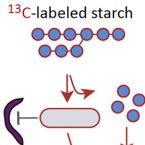
Abstract:
Humans and animals host diverse communities of microorganisms important to their physiology and health. Despite extensive sequencing-based characterization of host-associated microbiomes, there remains a dramatic lack of understanding of microbial functions. Stable-isotope probing (SIP) is a powerful strategy to elucidate the ecophysiology of microorganisms in complex host-associated microbiotas. Here, we suggest that SIP methodologies should be more frequently exploited as part of a holistic functional microbiomics approach. We provide examples of how SIP has been used to study host-associated microbes in vivo and ex vivo. We highlight recent developments in SIP technologies and discuss future directions that will facilitate deeper insights into the function of human and animal microbiomes.
Bacterial interactions during sequential degradation of cyanobacterial necromass in a sulfidic arctic marine sediment
2018 - Environ Microbiol, 20: 2927–2940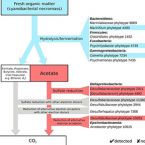
Abstract:
Seafloor microorganisms impact global carbon cycling by mineralizing vast quantities of organic matter (OM) from pelagic primary production, which is predicted to increase in the Arctic because of diminishing sea ice cover. We studied microbial interspecies-carbon-flow during anaerobic OM degradation in arctic marine sediment using stable isotope probing. We supplemented sediment incubations with 13C-labeled cyanobacterial necromass (spirulina), mimicking fresh OM input, or acetate, an important OM degradation intermediate, and monitored sulfate reduction rates and concentrations of volatile fatty acids (VFAs) during substrate degradation. Sequential 16S rRNA gene and transcript amplicon sequencing and fluorescence in situ hybridization combined with Raman microspectroscopy revealed that only few bacterial species were the main degraders of 13C-spirulina necromass. Psychrilyobacter, Psychromonas, Marinifilum, Colwellia, Marinilabiaceae and Clostridiales species were likely involved in the primary hydrolysis and fermentation of spirulina. VFAs, mainly acetate, produced from spirulina degradation were mineralized by sulfate-reducing bacteria and an Arcobacter species. Cellular activity of Desulfobacteraceae and Desulfobulbaceae species during acetoclastic sulfate reduction was largely decoupled from relative 16S rRNA gene abundance shifts. Our findings provide new insights into the identities and physiological constraints that determine the population dynamics of key microorganisms during complex OM degradation in arctic marine sediments.
Peatland Acidobacteria with a dissimilatory sulfur metabolism
2018 - ISME J, 12: 1729-1742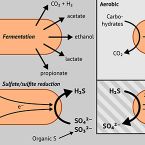
Abstract:
Sulfur-cycling microorganisms impact organic matter decomposition in wetlands and consequently greenhouse gas emissions from these globally relevant environments. However, their identities and physiological properties are largely unknown. By applying a functional metagenomics approach to an acidic peatland, we recovered draft genomes of seven novel Acidobacteria species with the potential for dissimilatory sulfite (dsrAB, dsrC, dsrD, dsrN, dsrT, dsrMKJOP) or sulfate respiration (sat, aprBA, qmoABC plus dsr genes). Surprisingly, the genomes also encoded DsrL, which so far was only found in sulfur-oxidizing microorganisms. Metatranscriptome analysis demonstrated expression of acidobacterial sulfur-metabolism genes in native peat soil and their upregulation in diverse anoxic microcosms. This indicated an active sulfate respiration pathway, which, however, might also operate in reverse for dissimilatory sulfur oxidation or disproportionation as proposed for the sulfur-oxidizing Desulfurivibrio alkaliphilus. Acidobacteria that only harbored genes for sulfite reduction additionally encoded enzymes that liberate sulfite from organosulfonates, which suggested organic sulfur compounds as complementary energy sources. Further metabolic potentials included polysaccharide hydrolysis and sugar utilization, aerobic respiration, several fermentative capabilities, and hydrogen oxidation. Our findings extend both, the known physiological and genetic properties of Acidobacteria and the known taxonomic diversity of microorganisms with a DsrAB-based sulfur metabolism, and highlight new fundamental niches for facultative anaerobic Acidobacteria in wetlands based on exploitation of inorganic and organic sulfur molecules for energy conservation.
Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle
2018 - ISME J, 12: 1715-1728
Abstract:
A critical step in the biogeochemical cycle of sulfur on Earth is microbial sulfate reduction, yet organisms from relatively few lineages have been implicated in this process. Previous studies using functional marker genes have detected abundant, novel dissimilatory sulfite reductases (DsrAB) that could confer the capacity for microbial sulfite/sulfate reduction but were not affiliated with known organisms. Thus, the identity of a significant fraction of sulfate/sulfite-reducing microbes has remained elusive. Here we report the discovery of the capacity for sulfate/sulfite reduction in the genomes of organisms from thirteen bacterial and archaeal phyla, thereby more than doubling the number of microbial phyla associated with this process. Eight of the thirteen newly identified groups are candidate phyla that lack isolated representatives, a finding only possible given genomes from metagenomes. Organisms from Verrucomicrobia and two candidate phyla, Candidatus Rokubacteria and Candidatus Hydrothermarchaeota, contain some of the earliest evolved dsrAB genes. The capacity for sulfite reduction has been laterally transferred in multiple events within some phyla, and a key gene potentially capable of modulating sulfur metabolism in associated cells has been acquired by putatively symbiotic bacteria. We conclude that current functional predictions based on phylogeny significantly underestimate the extent of sulfate/sulfite reduction across Earth’s ecosystems. Understanding the prevalence of this capacity is integral to interpreting the carbon cycle because sulfate reduction is often coupled to turnover of buried organic carbon. Our findings expand the diversity of microbial groups associated with sulfur transformations in the environment and motivate revision of biogeochemical process models based on microbial community composition.
Draft genome sequence of Telmatospirillum siberiense 26-4b1T, an acidotolerant peatland alphaproteobacterium potentially involved in sulfur cycling
2018 - Genome Announc, 6: e01524-17
Abstract:
The facultative anaerobic chemoorganoheterotrophic alphaproteobacterium Telmatospirillum siberiense 26-4b1T was isolated from a Siberian peatland. We report on a 6.20 Mbp near complete, high quality draft genome of T. siberiense that reveals expected and novel metabolic potential for the genus Telmatospirillum, including genes for sulfur oxidation.
The life sulfuric: microbial ecology of sulfur cycling in marine sediments.
2017 - Environ Microbiol Rep, 4: 323-344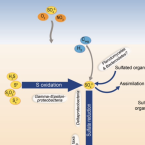
Abstract:
Almost the entire seafloor is covered with sediments that can be more than 10 000 m thick and represent a vast microbial ecosystem that is a major component of Earth's element and energy cycles. Notably, a significant proportion of microbial life in marine sediments can exploit energy conserved during transformations of sulfur compounds among different redox states. Sulfur cycling, which is primarily driven by sulfate reduction, is tightly interwoven with other important element cycles (carbon, nitrogen, iron, manganese) and therefore has profound implications for both cellular- and ecosystem-level processes. Sulfur-transforming microorganisms have evolved diverse genetic, metabolic, and in some cases, peculiar phenotypic features to fill an array of ecological niches in marine sediments. Here, we review recent and selected findings on the microbial guilds that are involved in the transformation of different sulfur compounds in marine sediments and emphasise how these are interlinked and have a major influence on ecology and biogeochemistry in the seafloor. Extraordinary discoveries have increased our knowledge on microbial sulfur cycling, mainly in sulfate-rich surface sediments, yet many questions remain regarding how sulfur redox processes may sustain the deep-subsurface biosphere and the impact of organic sulfur compounds on the marine sulfur cycle.
Ammonia-oxidising archaea living at low pH: Insights from comparative genomics.
2017 - Environ. Microbiol., 12: 4939-4952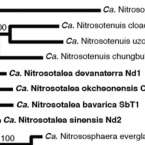
Abstract:
Obligate acidophilic members of the thaumarchaeotal genus Candidatus Nitrosotalea play an important role in nitrification in acidic soils, but their evolutionary and physiological adaptations to acidic environments are still poorly understood, with only a single member of this genus (Ca. N. devanaterra) having its genome sequenced. In this study, we sequenced the genomes of two additional cultured Ca. Nitrosotalea strains, extracted an almost complete Ca. Nitrosotalea metagenome-assembled genome from an acidic fen, and performed comparative genomics of the four Ca. Nitrosotalea genomes with 19 other archaeal ammonia oxidiser genomes. Average nucleotide and amino acid identities revealed that the four Ca. Nitrosotalea strains represent separate species within the genus. The four Ca. Nitrosotalea genomes contained a core set of 103 orthologous gene families absent from all other ammonia-oxidizing archaea and, for most of these gene families, expression could be demonstrated in laboratory culture or the environment via proteomic or metatranscriptomic analyses respectively. Phylogenetic analyses indicated that four of these core gene families were acquired by the Ca. Nitrosotalea common ancestor via horizontal gene transfer from acidophilic representatives of Euryarchaeota. We hypothesize that gene exchange with these acidophiles contributed to the competitive success of the Ca. Nitrosotalea lineage in acidic environments.
Bottled aqua incognita: Microbiota assembly and dissolved organic matter diversity in natural mineral waters
2017 - Microbiome, 5: 126
Abstract:
Background: Non-carbonated natural mineral waters contain microorganisms that regularly grow after bottling despite low concentrations of dissolved organic matter (DOM). Yet, the compositions of bottled water microbiota and organic substrates that fuel microbial activity, and how both change after bottling, are still largely unknown.
Results: We performed a multifaceted analysis of microbiota and DOM diversity in twelve natural mineral waters from six European countries. 16S rRNA gene-based analyses showed that less than ten species-level operational taxonomic units (OTUs) dominated the bacterial communities in the water phase and associated with the bottle wall after a short phase of post-bottling growth. Members of the betaproteobacterial genera Curvibacter, Aquabacterium, and Polaromonas (Comamonadaceae) grew in most waters and represent ubiquitous, mesophilic, heterotrophic aerobes in bottled waters. Ultrahigh-resolution mass spectrometry of DOM in bottled waters and their corresponding source waters identified thousands of molecular formulae characteristic of mostly refractory, soil-derived DOM.
Conclusions. The bottle environment, including source water physicochemistry, selected for growth of a similar low-diversity microbiota across various bottled waters. Relative abundance changes of hundreds of multi-carbon molecules were related to growth of less than ten abundant OTUs. We thus speculate that individual bacteria cope with oligotrophic conditions by simultaneously consuming diverse DOM molecules.
Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay
2017 - Appl Environ Microbiol, 83: e01547-17
Abstract:
Most sulfate-reducing microorganisms (SRM) present in subsurface marine sediments belong to uncultured groups only distantly related to known SRM and it remains unclear how changing geochemical zones and sediment depth influence their community structure. We mapped the community composition and abundance of SRM by amplicon-sequencing and quantifying dsrB, which encodes dissimilatory sulfite reductase subunit beta, in sediment samples covering different vertical geochemical zones ranging from the surface sediment to the deep sulfate-depleted subsurface at four locations in Aarhus Bay, Denmark. SRM were present in all geochemical zones including sulfate-depleted methanogenic sediment. The biggest shift in SRM community composition and abundance occurring across the transition from bioturbated surface sediments into non-bioturbated sediments below, where redox fluctuations and input of fresh organic matter due to macrofaunal activity are absent. SRM abundance correlated with sulfate reduction rates determined for the same sediments. Sulfate availability showed weaker correlation with SRM abundances and no significant correlation with the composition of the SRM community. The overall SRM species diversity decreased with depth, yet we identified a subset of highly abundant community members that persists across all vertical geochemical zones of all stations. We conclude that subsurface SRM communities assemble by persistence of members of the surface community and that the transition from the bioturbated surface sediment to the unmixed sediment below is a main site of assembly of the subsurface SRM community.
HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis
2017 - Cancer Res., 77: 2424-2438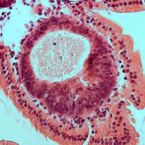
Abstract:
HuR is an RNA-binding protein implicated in immune homeostasis and various cancers, including colorectal cancer. HuR binding to AU-rich elements within the 3' untranslated region of mRNAs encoding oncogenes, growth factors, and various cytokines leads message stability and translation. In this study, we evaluated HuR as a small-molecule target for preventing colorectal cancer in high-risk groups such as those with familial adenomatosis polyposis (FAP) or inflammatory bowel disease (IBD). In human specimens, levels of cytoplasmic HuR were increased in colonic epithelial cells from patients with IBD, IBD-cancer, FAP-adenoma, and colorectal cancer, but not in patients with IBD-dysplasia. Intraperitoneal injection of the HuR small-molecule inhibitor MS-444 in AOM/DSS mice, a model of IBD and inflammatory colon cancer, augmented DSS-induced weight loss and increased tumor multiplicity, size, and invasiveness. MS-444 treatment also abrogated tumor cell apoptosis and depleted tumor-associated eosinophils, accompanied by a decrease in IL18 and eotaxin-1. In contrast, HuR inhibition in APCMin mice, a model of FAP and colon cancer, diminished the number of small intestinal tumors generated. In this setting, fecal microbiota, evaluated by 16S rRNA gene amplicon sequencing, shifted to a state of reduced bacterial diversity, with an increased representation of Prevotella, Akkermansia, and Lachnospiraceae Taken together, our results indicate that HuR activation is an early event in FAP-adenoma but is not present in IBD-dysplasia. Furthermore, our results offer a preclinical proof of concept for HuR inhibition as an effective means of FAP chemoprevention, with caution advised in the setting of IBD.
Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota
2017 - mSystems, 2: e00171-16
Abstract:
Mucispirillum schaedleri is an abundant inhabitant of the intestinal mucus layer of rodents and other animals and has been suggested to be a pathobiont, a commensal that plays a role in disease. In order to gain insights into its lifestyle, we analyzed the genome and transcriptome of M. schaedleri ASF 457 and performed physiological experiments to test traits predicted by its genome. Although described as a mucus inhabitant, M. schaedleri has limited capacity for degrading host-derived mucosal glycans and other complex polysaccharides. Additionally, M. schaedleri reduces nitrate and expresses systems for scavenging oxygen and reactive oxygen species in vivo, which may account for its localization close to the mucosal tissue and expansion during inflammation. Also of note, M. schaedleri harbors a type VI secretion system and putative effector proteins and can modify gene expression in mucosal tissue, suggesting intimate interactions with its host and a possible role in inflammation. The M. schaedleri genome has been shaped by extensive horizontal gene transfer, primarily from intestinal Epsilon- and Deltaproteobacteria, indicating that horizontal gene transfer has played a key role in defining its niche in the gut ecosystem.
Stable isotope techniques for the assessment of host and microbiota response during gastrointestinal dysfunction
2017 - J Pediatr Gastroenterol Nutr, 64: 8-14
Abstract:
The International Atomic Energy Agency convened a technical meeting on environmental enteric dysfunction (EED) in Vienna (28th – 30th October 2015; https://nucleus.iaea.org/HHW/Nutrition/EED_Technical_Meeting/index.html) to bring together international experts in the fields of EED, nutrition and stable isotope technologies. Advances in stable isotope labelling techniques open up new possibilities to improve our understanding of gastrointestinal dysfunction and the role of the microbiota in host health. In the context of EED, little is known about the role gut dysfunction may play in macro- and micronutrient bioavailability and requirements and what the consequences may be for nutritional status and linear growth. Stable isotope labelling techniques have been used to assess intestinal mucosal injury and barrier function, carbohydrate digestion and fermentation, protein derived amino acid bioavailability and requirements, micronutrient bioavailability and to track microbe-microbe and microbe-host interactions at the single cell level. The non-invasive nature of stable isotope technologies potentially allows for low-hazard, field deployable tests of gut dysfunction that are applicable across all age-groups. The purpose of this review is to assess the state-of-the-art in the use of stable isotope technologies and to provide a perspective on where these technologies can be exploited to further our understanding of gut dysfunction in EED.
Genome-guided design of a novel defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium
2016 - Nature Microbiol, 2: 16215
Abstract:
Protection against enteric infections, also termed colonization resistance, results from mutualistic interactions of the host and its indigenous microbes. The gut microbiota of humans and mice is highly diverse and it is therefore challenging to assign specific properties to its individual members. Here, we have used a collection of murine bacterial strains and a modular design approach to create a minimal bacterial community that, once established in germ-free mice, provided colonization resistance against the human enteric pathogen Salmonella enterica serovar Typhimurium (S. Tm). Initially, a community of 12 strains, termed Oligo-Mouse Microbiota (Oligo-MM12), representing members of the major bacterial phyla in the murine gut, was selected. This community was stable over consecutive mouse generations and provided colonization resistance against S. Tm infection, albeit not to the degree of a conventional complex microbiota. Comparative (meta)genome analyses identified functions represented in a conventional microbiome but absent from the Oligo-MM12. By genome-informed design, we created an improved version of the Oligo-MM community harbouring three facultative anaerobic bacteria from the Mouse Intestinal Bacterial Collection (miBC) that provided conventional-like colonization resistance. In conclusion, we have established a highly versatile experimental system that showed efficacy in an enteric infection model. Thus, in combination with exhaustive bacterial strain collections and systems-based approaches, genomeguided design can be used to generate insights into microbe–microbe and microbe–host interactions for the investigation of ecological and disease-relevant mechanisms in the intestine.
Environmental enteric dysfunction and growth failure/stunting in global child health
2016 - Pediatrics, 138: e20160641
Abstract:
Approximately 25% of the world’s children under age 5 years have stunted growth, which is associated with increased mortality, cognitive dysfunction, and loss of productivity. Reducing by 40% the number of stunted children is a global target for 2030. The pathogenesis of stunting is poorly understood. Pre- and post-natal nutritional deficits and enteric and systemic infections clearly contribute, but recent findings implicate a central role for environmental enteric dysfunction (EED), a generalized disturbance of small intestinal structure and function found at high prevalence in children living under unsanitary conditions. Mechanisms contributing to growth failure in EED include intestinal leakiness and heightened permeability, gut inflammation, dysbiosis and bacterial translocation, systemic inflammation, and nutrient malabsorption. Since EED has multiple causal pathways, approaches to manage it need to be multi-faceted. Potential interventions to tackle EED include: a) reduction of exposure to feces and contact with animals through programs like improved water, sanitation and hygiene (WASH); b) breastfeeding and enhanced dietary diversity; c) probiotics and prebiotics; d) nutrient supplements including zinc, polyunsaturated fatty acids, and amino acids; e) anti-inflammatory agents such as 5-aminosalicyclic acid; and f) antibiotics in the context of acute malnutrition and infection. Better understanding of the underlying causes of EED, and development of non-invasive, practical, simple, and affordable point of care diagnostic tools remain key gaps. ‘Omics’ technologies (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) and stable isotope techniques (e.g., 13Carbon breath tests) targeted at children and their intestinal microbiota will enhance our ability to successfully identify, manage, and prevent the disorder.
Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses
2016 - Nature, 537: 689–693
Abstract:
Ocean microbes drive biogeochemical cycling on a global scale1. However, this cycling is constrained by viruses that affect community composition, metabolic activity, and evolutionary trajectories2, 3. Owing to challenges with the sampling and cultivation of viruses, genome-level viral diversity remains poorly described and grossly understudied, with less than 1% of observed surface-ocean viruses known4. Here we assemble complete genomes and large genomic fragments from both surface- and deep-ocean viruses sampled during the Tara Oceans and Malaspina research expeditions5, 6, and analyse the resulting ‘global ocean virome’ dataset to present a global map of abundant, double-stranded DNA viruses complete with genomic and ecological contexts. A total of 15,222 epipelagic and mesopelagic viral populations were identified, comprising 867 viral clusters (defined as approximately genus-level groups7, 8). This roughly triples the number of known ocean viral populations4 and doubles the number of candidate bacterial and archaeal virus genera8, providing a near-complete sampling of epipelagic communities at both the population and viral-cluster level. We found that 38 of the 867 viral clusters were locally or globally abundant, together accounting for nearly half of the viral populations in any global ocean virome sample. While two-thirds of these clusters represent newly described viruses lacking any cultivated representative, most could be computationally linked to dominant, ecologically relevant microbial hosts. Moreover, we identified 243 viral-encoded auxiliary metabolic genes, of which only 95 were previously known. Deeper analyses of four of these auxiliary metabolic genes (dsrC, soxYZ, P-II (also known as glnB) and amoC) revealed that abundant viruses may directly manipulate sulfur and nitrogen cycling throughout the epipelagic ocean. This viral catalog and functional analyses provide a necessary foundation for the meaningful integration of viruses into ecosystem models where they act as key players in nutrient cycling and trophic networks.
Bacterial nutrient foraging in a mouse model of enteral nutrient deprivation: Insight into the gut origin of sepsis
2016 - Am J Physiol Gastrointest Liver Physiol, 311: G734-G743
Abstract:
Total parenteral nutrition (TPN) leads to a shift in small intestinal microbiota with a characteristic dominance of Proteobacteria. This study examined how metabolomic changes within the small bowel support an altered microbial community in enterally deprived mice.
C57BL/6 mice were given TPN or enteral chow. Metabolomic analysis of jejunal contents was performed by liquid chromatography/mass spectrometry (LC/MS). In some experiments, leucine in TPN was partly substituted with (13)C-leucine. Additionally, jejunal contents from TPN dependent and enterally fed mice were gavaged into germ-free mice to reveal if the TPN phenotype was transferrable.
Small bowel contents of TPN mice maintained an amino acid composition similar to that of the TPN solution. Mass spectrometry analysis of small bowel contents of TPN dependent mice showed increased concentration of (13)C compared to fed mice receiving saline enriched with (13)C-leucine. (13)C-leucine added to the serosal side of Ussing chambers showed rapid permeation across TPN-dependent jejunum, suggesting increased transmucosal passage. Single-cell analysis by fluorescence in situ hybridization (FISH) - NanoSIMS demonstrated uptake of (13)C-leucine by TPN-associated bacteria, with preferential uptake by Enterobacteriaceae. Gavage of small bowel effluent from TPN mice into germ-free, fed mice resulted in a trend toward the pro-inflammatory TPN-phenotype with loss of epithelial barrier function.
TPN-dependence leads to increased permeation of TPN-derived nutrients into the small intestinal lumen, where they are predominately utilized by Enterobacteriaceae. The altered metabolomic composition of the intestinal lumen during TPN promotes dysbiosis.Single cell genome and group-specific dsrAB sequencing implicate members of the class Dehalococcoidia (phylum Chloroflexi) in sulfur cycling
2016 - mBio, 7: e00266-16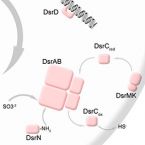
Abstract:
The marine subsurface sediment biosphere is widely inhabited by bacteria affiliated with the class Dehalococcoidia (DEH), phylum Chloroflexi, yet little is known regarding their metabolisms. In this report, genomic content from a single DEH cell (‘DEH-C11’) with a 16S rRNA gene that affiliated with a diverse cluster of 16S rRNA gene sequences prevalent in marine sediments, was obtained from sediments of Aarhus Bay, Denmark. The distinctive gene content of this cell suggests metabolic characteristics that differ from those of known DEH and Chloroflexi. Genes encoding dissimilatory sulfite reductase (Dsr) suggest DEH could respire oxidized sulfur compounds, although Chloroflexi have never been implicated in this mode of sulfur cycling. Using long-range PCR assays targeting DEH dsr-loci, dsrAB were amplified and sequenced from various marine sediments. Many of the amplified dsrAB sequences affiliated with the DEH Dsr-clade, which we propose equates to a family-level clade. This provides supporting evidence for the potential for sulfite reduction by diverse DEH. DEH-C11 also harboured genes encoding reductases for arsenate, dimethyl sulfoxide and halogenated organics. The reductive dehalogenase homolog (RdhA) forms a monophyletic clade along with RdhA sequences from various DEH-derived contigs retrieved from available metagenomes. Multiple facts indicate this RdhA may not be a terminal reductase. Other genes indicated nutrients and energy may be derived from the oxidation of substituted homocyclic and heterocyclic aromatic compounds. Together, these results suggest that marine DEH play a previously unrecognised role in sulfur cycling, and reveal potential for expanded catabolic and respiratory functions among subsurface DEH.
Gypsum amendment to rice paddy soil stimulated bacteria involved in sulfur cycling but largely preserved the phylogenetic composition of the total bacterial community
2016 - Environ Microbiol Rep, 8: 413-23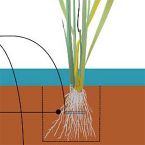
Abstract:
Rice paddies are indispensable for human food supply but emit large amounts of the greenhouse gas methane. Sulfur cycling occurs at high rates in these water-submerged soils and controls methane production, an effect that is increased by sulfate-containing fertilizers or soil amendments. We grew rice plants until their late vegetative phase with and without gypsum (CaSO4 ·2H2 O) amendment and identified responsive bacteria by 16S rRNA gene amplicon sequencing. Gypsum amendment decreased methane emissions by up to 99% but had no major impact on the general phylogenetic composition of the bacterial community. It rather selectively stimulated or repressed a small number of 129 and 27 species-level operational taxonomic units (OTUs) (out of 1,883-2,287 observed) in the rhizosphere and bulk soil, respectively. Gypsum-stimulated OTUs were affiliated with several potential sulfate-reducing (Syntrophobacter, Desulfovibrio, unclassified Desulfobulbaceae, unclassified Desulfobacteraceae) and sulfur-oxidizing taxa (Thiobacillus, unclassified Rhodocyclaceae), while gypsum-repressed OTUs were dominated by aerobic methanotrophs (Methylococcaceae). Abundance correlation networks suggested that two abundant (>1%) OTUs (Desulfobulbaceae, Rhodocyclaceae) were central to the reductive and oxidative parts of the sulfur cycle.
Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms
2016 - ISME J, 10: 2365-75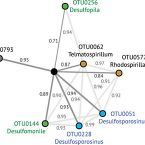
Abstract:
Dissimilatory sulfate reduction in peatlands is sustained by a cryptic sulfur cycle and effectively competes with methanogenic degradation pathways. In a series of peat soil microcosms incubated over 50 days, we identified bacterial consortia that responded to small, periodic additions of individual fermentation products (formate, acetate, propionate, lactate or butyrate) in the presence or absence of sulfate. Under sulfate supplementation, net sulfate turnover (ST) steadily increased to 16-174 nmol cm-3 per day and almost completely blocked methanogenesis. 16S rRNA gene and cDNA amplicon sequencing identified microorganisms whose increases in ribosome numbers strongly correlated to ST. Natively abundant (⩾0.1% estimated genome abundance) species-level operational taxonomic units (OTUs) showed no significant response to sulfate. In contrast, low-abundance OTUs responded significantly to sulfate in incubations with propionate, lactate and butyrate. These OTUs included members of recognized sulfate-reducing taxa (Desulfosporosinus, Desulfopila, Desulfomonile, Desulfovibrio) and also members of taxa that are either yet unknown sulfate reducers or metabolic interaction partners thereof. Most responsive OTUs markedly increased their ribosome content but only weakly increased in abundance. Responsive Desulfosporosinus OTUs even maintained a constantly low population size throughout 50 days, which suggests a novel strategy of rare biosphere members to display activity. Interestingly, two OTUs of the non-sulfate-reducing genus Telmatospirillum (Alphaproteobacteria) showed strongly contrasting preferences towards sulfate in butyrate-amended microcosms, corroborating that closely related microorganisms are not necessarily ecologically coherent. We show that diverse consortia of low-abundance microorganisms can perform peat soil sulfate reduction, a process that exerts control on methane production in these climate-relevant ecosystems.
Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics
2016 - Environ Microbiol, 18: 2994-3009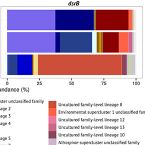
Abstract:
Genes encoding dissimilatory sulfite reductase (DsrAB) are commonly used as diagnostic markers in ecological studies of sulfite- and sulfate-reducing microorganisms. Here, we developed new high-coverage primer sets for generation of reductive bacterial-type dsrA and dsrB PCR products for highly parallel amplicon sequencing and a bioinformatics workflow for processing and taxonomic classification of short dsrA and dsrB reads. We employed two diverse mock communities that consisted of 45 or 90 known dsrAB sequences derived from environmental clones to precisely evaluate the performance of individual steps of our amplicon sequencing approach on the Illumina MiSeq platform. Although PCR cycle number, gene-specific primer mismatches, and stringent filtering for high-quality sequences had notable effects on the observed dsrA and dsrB community structures, recovery of most mock community sequences was generally proportional to their relative input abundances. Successful dsrA and dsrB diversity analysis in selected environmental samples further proved that the multiplex amplicon sequencing approach is adequate for monitoring spatial distribution and temporal abundance dynamics of dsrAB-containing microorganisms. While tested for reductive bacterial-type dsrAB, this method is readily applicable for oxidative-type dsrAB of sulfur-oxidizing bacteria and also provides guidance for processing short amplicon reads of other functional genes.
probeBase - an online resource for rRNA-targeted oligonucleotide probes and primers: new features 2016
2016 - Nucleic Acids Res, 44: D586-D589
Abstract:
probeBase http://www.probebase.net is a manually maintained and curated database of rRNA-targeted oligonucleotide probes and primers. Contextual information and multiple options for evaluating in silico hybridization performance against the most recent rRNA sequence databases are provided for each oligonucleotide entry, which makes probeBase an important and frequently used resource for microbiology research and diagnostics. Here we present a major update of probeBase, which was last featured in the NAR Database Issue 2007. This update describes a complete remodeling of the database architecture and environment to accommodate computationally efficient access. Improved search functions, sequence match tools, and data output now extend the opportunities for finding suitable hierarchical probe sets that target an organism or taxon at different taxonomic levels. To facilitate the identification of complementary probe sets for organisms represented by short rRNA sequence reads generated by amplicon sequencing or metagenomic analysis with next generation sequencing technologies such as Illumina and IonTorrent, we introduce a novel tool that recovers surrogate near full-length rRNA sequences for short query sequences and finds matching oligonucleotides in probeBase.
Phylogenetic and genomic analysis of Methanomassiliicoccales in wetlands and animal intestinal tracts reveals clade-specific habitat preferences
2016 - FEMS Microbiol Ecol, 92: fiv149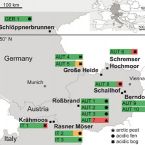
Abstract:
Methanogenic Thermoplasmata of the novel order Methanomassiliicoccales were recently discovered in human and animal gastro-intestinal tracts (GITs). However their distribution in other methanogenic environments has not been addressed systematically. Here we surveyed Methanomassiliicoccales presence in wetland soils, a globally important source of methane emissions to the atmosphere, and in the GITs of different animals by PCR targeting their 16S rRNA and methyl:coenzyme M reductase (α-subunit) genes. We detected Methanomassiliicoccales in all 16 peat soils investigated, indicating their wide distribution in these habitats. Additionally, we detected their genes in various animal feces. Methanomassiliicoccales were subdivided in two broad phylogenetic clades designated 'environmental' and 'GIT' clades based on differential, although non-exclusive, habitat preferences of their members. A well-supported cluster within the environmental clade comprised more than 80% of all wetland 16S rRNA gene sequences. Metagenome assembly from bovine rumen fluid enrichments resulted in two almost complete genomes of both Methanomassiliicoccales clades. Comparative genomics revealed that members of the environmental clade contain larger genomes and a higher number of genes encoding anti-oxidative enzymes than animal GIT clade representatives. This study highlights the wide distribution of Methanomassiliicoccales in wetlands, which suggests that they contribute to methane emissions from these climate-relevant ecosystems.
Activity and community structures of sulfate-reducing microorganisms in polar, temperate and tropical marine sediments
2016 - ISME J, 10: 796–809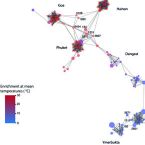
Abstract:
Temperature has a fundamental impact on the metabolic rates of microorganisms and strongly influences microbial ecology and biogeochemical cycling in the environment. In this study, we examined the catabolic temperature response of natural communities of sulfate-reducing microorganisms (SRM) in polar, temperate, and tropical marine sediments. In short-term sediment incubation experiments with 35S-sulfate, we demonstrated how the cardinal temperatures for sulfate reduction correlate with mean annual sediment temperatures, indicating specific thermal adaptations of the dominant SRM in each of the investigated ecosystems. The community structure of putative SRM in the sediments, as revealed by pyrosequencing of bacterial 16S rRNA gene amplicons and phylogenetic assignment to known SRM taxa, consistently correlated with in situ temperatures, but not with sediment organic carbon concentrations or C:N ratios of organic matter. Additionally, several species-level SRM phylotypes of the class Deltaproteobacteria tended to co-occur at sites with similar mean annual temperatures, regardless of geographic distance. The observed temperature adaptations of SRM imply that environmental temperature is a major controlling variable for physiological selection and ecological and evolutionary differentiation of microbial communities.
Intestinal microbiota signatures associated with inflammation history in mice experiencing recurring colitis
2015 - Front Microbiol, 6: 1408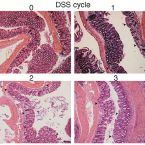
Abstract:
Acute colitis causes alterations in the intestinal microbiota, but the microbiota is thought to recover after such events. Extreme microbiota alterations are characteristic of human chronic inflammatory bowel diseases, although alterations reported in different studies are divergent and sometimes even contradictory. To better understand the impact of periodic disturbances on the intestinal microbiota and its compositional difference between acute and relapsing colitis, we investigated the beginnings of recurrent inflammation using the dextran sodium sulfate (DSS) mouse model of chemically induced colitis. Using bacterial 16S rRNA gene-targeted pyrosequencing as well as quantitative fluorescence in situ hybridization, we profiled the intestinal and stool microbiota of mice over the course of three rounds of DSS-induced colitis and recovery. We found that characteristic inflammation-associated microbiota could be detected in recovery-phase mice. Successive inflammation episodes further drove the microbiota into an increasingly altered composition post-inflammation, and signatures of colitis history were detectable in the microbiota more sensitively than by pathology analysis. Bacterial indicators of murine colitis history were identified in intestinal and stool samples, with a high degree of consistency between both sample types. Stool may therefore be a promising non-invasive source of bacterial biomarkers that are highly sensitive to inflammation state and history.
Intestinal epithelial cell tyrosine kinase 2 transduces interleukin-22 signals to protect from acute colitis
2015 - J Immunol., 195: 5011-5024
Abstract:
In the intestinal tract, IL-22 activates signal transducer and activator of transcription 3 (STAT3) to promote intestinal epithelial cell (IEC) homeostasis and tissue healing. The mechanism has remained obscure but we demonstrate that IL-22 acts via tyrosine kinase 2 (Tyk2), a member of the Janus kinase (Jak) family. Using a mouse model for colitis, we show that Tyk2 deficiency is associated with an altered composition of the gut microbiota and exacerbates inflammatory bowel disease (IBD). Colitic Tyk2-/- mice have less phosphorylated STAT3 (pY-STAT3) in colon tissue and their IECs proliferate less efficiently. Tyk2-deficient primary IECs show reduced pY-STAT3 in response to IL-22 stimulation and expression of IL-22-STAT3 target genes is reduced in IECs from healthy and colitic Tyk2-/- mice. Experiments with conditional Tyk2-/- mice reveal that IEC-specific depletion of Tyk2 aggravates colitis. Disease symptoms can be alleviated by administering high doses of recombinant IL-22-Fc, indicating that Tyk2 deficiency can be rescued via the IL-22 receptor complex. The pivotal function of Tyk2 in IL-22-dependent colitis was confirmed in Citrobacter rodentium-induced disease. Thus, Tyk2 protects against acute colitis in part by amplifying inflammation-induced epithelial IL-22 signaling to STAT3.
A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes.
2015 - Front Microbiol, 6: 731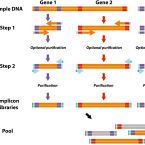
Abstract:
High throughput sequencing of phylogenetic and functional gene amplicons provides tremendous insight into the structure and functional potential of complex microbial communities. Here, we introduce a highly adaptable and economical PCR approach to barcoding and pooling libraries of numerous target genes. In this approach, we replace gene- and sequencing platform-specific fusion primers with general, interchangeable barcoding primers, enabling nearly limitless customized barcode-primer combinations. Compared to barcoding with long fusion primers, our multiple-target gene approach is more economical because it overall requires lower number of primers and is based on short primers with generally lower synthesis and purification costs. To highlight our approach, we pooled over 900 different small-subunit rRNA and functional gene amplicon libraries obtained from various environmental or host-associated microbial community samples into a single, paired-end Illumina MiSeq run. Although the amplicon regions ranged in size from approximately 290 to 720 bp, we found no significant systematic sequencing bias related to amplicon length or gene target. Our results indicate that this flexible multiplexing approach produces large, diverse, and high quality sets of amplicon sequence data for modern studies in microbial ecology.
Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases
2015 - ISME J, 9: 1152-1165
Abstract:
The energy metabolism of essential microbial guilds in the biogeochemical sulfur cycle is based on a DsrAB-type dissimilatory (bi)sulfite reductase that either catalyzes the reduction of sulfite to sulfide during anaerobic respiration of sulfate, sulfite and organosulfonates, or acts in reverse during sulfur oxidation. Common use of dsrAB as a functional marker showed that dsrAB richness in many environments is dominated by novel sequence variants and collectively represents an extensive, largely uncharted sequence assemblage. Here, we established a comprehensive, manually curated dsrAB/DsrAB database and used it to categorize the known dsrAB diversity, reanalyze the evolutionary history of dsrAB and evaluate the coverage of published dsrAB-targeted primers. Based on a DsrAB consensus phylogeny, we introduce an operational classification system for environmentaldsrAB sequences that integrates established taxonomic groups with operational taxonomic units (OTUs) at multiple phylogenetic levels, ranging from DsrAB enzyme families that reflect reductive or oxidative DsrAB types of bacterial or archaeal origin, superclusters, uncultured family-level lineages to species-level OTUs. Environmental dsrAB sequences constituted at least 13 stable family-level lineages without any cultivated representatives, suggesting that major taxa of sulfite/sulfate-reducing microorganisms have not yet been identified. Three of these uncultured lineages occur mainly in marine environments, while specific habitat preferences are not evident for members of the other 10 uncultured lineages. In summary, our publically available dsrAB/DsrAB database, the phylogenetic framework, the multilevel classification system and a set of recommended primers provide a necessary foundation for large-scale dsrAB ecology studies with next-generation sequencing methods.
Functionally relevant diversity of closely related Nitrospira in activated sludge
2015 - ISME J, 9: 643-655
Abstract:
Nitrospira are chemolithoautotrophic nitrite-oxidizing bacteria that catalyze the second step of nitrification in most oxic habitats and are important for excess nitrogen removal from sewage in wastewater treatment plants (WWTPs). To date, little is known about their diversity and ecological niche partitioning within complex communities. In this study, the fine-scale community structure and function of Nitrospira was analyzed in two full-scale WWTPs as model ecosystems. In Nitrospira-specific 16S rRNA clone libraries retrieved from each plant, closely related phylogenetic clusters (16S rRNA identities between clusters ranged from 95.8% to 99.6%) within Nitrospira lineages I and II were found. Newly designed probes for fluorescence in situ hybridization (FISH) allowed the specific detection of several of these clusters, whose coexistence in the WWTPs was shown for prolonged periods of several years. In situ ecophysiological analyses based on FISH, relative abundance and spatial arrangement quantification, as well as microautoradiography revealed functional differences of these Nitrospira clusters regarding the preferred nitrite concentration, the utilization of formate as substrate and the spatial coaggregation with ammonia-oxidizing bacteria as symbiotic partners. Amplicon pyrosequencing of the nxrB gene, which encodes subunit beta of nitrite oxidoreductase of Nitrospira, revealed in one of the WWTPs as many as 121 species-level nxrB operational taxonomic units with highly uneven relative abundances in the amplicon library. These results show a previously unrecognized highdiversity of Nitrospira in engineered systems, which is at least partially linked to niche differentiation and may have important implications for process stability.
Type I interferons have opposing effects during the emergence and recovery phases of colitis.
2014 - Eur J Immunol., 44: 2749-60
Abstract:
The contribution of the innate immune system to inflammatory bowel disease (IBD) is under intensive investigation. Research in animal models has demonstrated that type I interferons (IFN-Is) protect from IBD. In contrast, studies of patients with IBD have produced conflicting results concerning the therapeutic potential of IFN-Is. Here, we present data suggesting that IFN-Is play dual roles as regulators of intestinal inflammation in dextran sodium sulfate (DSS)-treated C57BL/6 mice. Though IFN-Is reduced acute intestinal damage and the abundance ofcolitis-associated intestinal bacteria caused by treatment with a high dose of DSS, they also inhibited the resolution of inflammation after DSS treatment. IFN-Is played an anti-inflammatory role by suppressing the release of IL-1β from the colon MHC class II(+) cells. Consistently, IL-1 receptor blockade reduced the severity of inflammation in IFN-I receptor-deficient mice and myeloid cell-restricted ablation of the IFN-I receptor was detrimental. The proinflammatory role of IFN-Is during recovery from DSS treatment was caused by IFN-I-dependent cell apoptosis as well as an increase in chemokine production and infiltrating inflammatory monocytes and neutrophils. Thus, IFN-Is play opposing roles in specificphases of intestinal injury and inflammation, which may be important for guiding treatment strategies in patients.
Endospores of thermophilic bacteria as tracers of microbial dispersal by ocean currents
2014 - ISME J., 8: 1153-65
Abstract:
Microbial biogeography is influenced by the combined effects of passive dispersal and environmental selection, but the contribution of either factor can be difficult to discern. As thermophilic bacteria cannot grow in the cold seabed, their inactive spores are not subject to environmental selection. We therefore conducted a global experimental survey using thermophilic endospores that are passively deposited by sedimentation to the cold seafloor as tracers to study the effect of dispersal by ocean currents on the biogeography of marine microorganisms. Our analysis of 81 different marine sediments from around the world identified 146 species-level 16S rRNA phylotypes of endospore-forming, thermophilic Firmicutes. Phylotypes showed various patterns of spatial distribution in the world oceans and were dispersal-limited to different degrees. Co-occurrence of several phylotypes in locations separated by great distances (west of Svalbard, the Baltic Sea and the Gulf of California) demonstrated a widespread but not ubiquitous distribution. In contrast, Arctic regions with water masses that are relatively isolated from global ocean circulation (Baffin Bay and east of Svalbard) were characterized by low phylotype richness and different compositions of phylotypes. The observed distribution pattern ofthermophilic endospores in marine sediments suggests that the impact of passive dispersal on marine microbial biogeography is controlled by the connectivity of local water masses to ocean circulation.
Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery
2014 - ISME J., 8(5):1101-14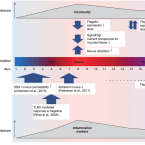
Abstract:
Although alterations in gut microbiota composition during acute colitis have been repeatedly observed, associated functional changes and therecovery from dysbiosis received little attention. In this study, we investigated structure and function of the gut microbiota during acute inflammationand recovery in a dextran sodium sulfate (DSS)-colitis mouse model using metatranscriptomics, bacterial 16S rRNA gene amplicon sequencing and monitoring of selected host markers. Parallel to an increase of host markers of inflammation during acute colitis, we observed relative abundance shifts and alterations in phylotype composition of the dominant bacterial orders Clostridiales and Bacteroidales, and an increase of the low abundant Enterobacteriales, Deferribacterales, Verrucomicrobiales and Erysipelotrichales. During recovery, the microbiota began to resume, but did not reach its original composition until the end of the experiment. Microbial gene expression was more resilient to disturbance, with pre-perturbation-type transcript profiles appearing quickly after acute colitis. The decrease of Clostridiales during inflammation correlated with a reduction of transcripts related to butyrate formation, suggesting a disturbance in host-microbe signalling and mucosal nutrient provision. The impact of acute inflammationon the Clostridiales was also characterized by a significant downregulation of their flagellin-encoding genes. In contrast, the abundance of members of the Bacteroidales increased along with an increase in transcripts related to mucin degradation. We propose that acute inflammation triggered a selective reaction of the immune system against flagella of commensals and temporarily altered murine microbiota composition and functions relevant for the host. Despite changes in specific interactions, the host-microbiota homeostasis revealed a remarkable ability for recovery.
NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira
2014 - Environ Microbiol, 16: 3055-3071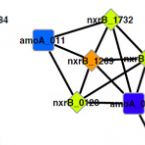
Abstract:
Nitrospira are the most widespread and diverse known nitrite-oxidizing bacteria and key nitrifiers in natural and engineered ecosystems. Nevertheless, their ecophysiology and environmental distribution are understudied due to the recalcitrance of Nitrospira to cultivation and the lack of a molecular functional marker, which would allow the detection of Nitrospira in the environment. Here we introduce nxrB, the gene encoding subunit beta of nitrite oxidoreductase, as a functional and phylogenetic marker for Nitrospira. Phylogenetic trees based on nxrB of Nitrospira were largely congruent to 16S rRNA-based phylogenies. By using new nxrB-selective PCR primers, we obtained almost full-length nxrB sequences from Nitrospira cultures, two activated sludge samples, and several geographically and climatically distinct soils. Amplicon pyrosequencing of nxrB fragments from 16 soils revealed a previously unrecognized diversity of terrestrial Nitrospira with 1,801 detected species-level OTUs (using an inferred species threshold of 95% nxrB identity). Richness estimates ranged from 10 to 946 co-existing Nitrospira species per soil. Comparison to an archaeal amoA dataset obtained from the same soils [Environ. Microbiol. 14: 525-539 (2012)] uncovered that ammonia-oxidizing archaea and Nitrospira communities were highly correlated across the soil samples, possibly indicating shared habitat preferences or specific biological interactions among members of these nitrifier groups.
Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation
2013 - Am. J. Gastroenterol., 108: 1620-1630
Abstract:
OBJECTIVES: Fecal microbiota transplantation (FMT) from healthy donors, which is an effective alternative for treatment of Clostridium difficile-associated disease, is being considered for several disorders such as inflammatory bowel disease, irritable bowel syndrome, and metabolic syndrome. Disease remission upon FMT is thought to be facilitated by an efficient colonization of healthy donor microbiota, but knowledge of the composition and temporal stability of patient microbiota after FMT is lacking.METHODS:Five patients with moderately to severely active ulcerative colitis (Mayo score ≥6) and refractory to standard therapy received FMT via nasojejunal tube and enema. In addition to clinical activity and adverse events, the patients' fecal bacterial communities were monitored at multiple time points for up to 12 weeks using 16S rRNA gene-targeted pyrosequencing. RESULTS:FMT elicited fever and a temporary increase of C-reactive protein. Abundant bacteria from donors established in recipients, but the efficiency and stability of donor microbiota colonization varied greatly. A positive clinical response was observed after 12 weeks in one patient whose microbiota had been effectively augmented by FMT. This augmentation was marked by successive colonization of donor-derived phylotypes including the anti-inflammatory and/or short-chain fatty acid-producing Faecalibacterium prausnitzii, Rosebura faecis, and Bacteroides ovatus. Disease severity (as measured by the Mayo score) was associated with an overrepresentation of Enterobacteriaceae and an underrepresentation of Lachnospiraceae. CONCLUSIONS:This study highlights the value of characterizing temporally resolved microbiota dynamics for a better understanding of FMT efficacy and provides potentially useful diagnostic indicators for monitoring FMT success in the treatment of ulcerative colitis.
Intestinal bacteria modify lymphoma incidence and latency by affecting systemic inflammatory state, oxidative stress, and leucocyte genotoxicity
2013 - Cancer Res., 73: 4222-4232
Abstract:
Ataxia-telangiectasia is a genetic disorder associated with high incidence of B-cell lymphoma. Using an ataxia-telangiectasia mouse model, we compared lymphoma incidence in several isogenic mouse colonies harboring different bacterial communities, finding that intestinal microbiota are a major contributor to disease penetrance and latency, lifespan, molecular oxidative stress, and systemic leukocyte genotoxicity. High-throughput sequence analysis of rRNA genes identified mucosa-associated bacterial phylotypes that were colony-specific. Lactobacillus johnsonii, which was deficient in the more cancer-prone mouse colony, was causally tested for its capacity to confer reduced genotoxicity when restored by short-term oral transfer. This intervention decreased systemic genotoxicity, a response associated with reduced basal leukocytes and the cytokine-mediated inflammatory state, and mechanistically linked to the host cell biology of systemic genotoxicity. Our results suggest that intestinal microbiota are a potentially modifiable trait for translational intervention in individuals at risk for B-cell lymphoma, or for other diseases that are driven by genotoxicity or the molecular response to oxidative stress.
Colonization resistance and microbial ecophysiology: Using gnotobiotic mouse models and single-cell technology to explore the intestinal jungle
2013 - FEMS Microbiol. Rev., 37: 793-829
Abstract:
The highly diverse intestinal microbiota forms a structured community engaged in constant communication with itself and its host and is characterized by extensive ecological interactions. A key benefit that the microbiota affords its host is its ability to protect against infections in a process termed colonization resistance (CR), which remains insufficiently understood. In this review, we connect basic concepts of CR with new insights from recent years and highlight key technological advances in the field of microbial ecology. We present a selection of statistical and bioinformatics tools used to generate hypotheses about synergistic and antagonistic interactions in microbial ecosystems from metagenomic datasets. We emphasize the importance of experimentally testing these hypotheses and discuss the value of gnotobiotic mouse models for investigating specific aspects related to microbiota-host-pathogen interactions in a well-defined experimental system. We further introduce new developments in the area of single-cell analysis using fluorescence in situ hybridization in combination with metabolic stable isotope labeling technologies for studying the in vivo activities of complex community members. These approaches promise to yield novel insights into the mechanisms of CR and intestinal ecophysiology in general, and give researchers the means to experimentally test hypotheses in vivo at varying levels of biological and ecological complexity.
Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing
2013 - Proc. Natl. Acad. Sci. USA, 110: 4720-4725
Abstract:
The animal and human intestinal mucosa secretes an assortment of compounds to establish a physical barrier between the host tissue and intestinal contents, a separation that is vital for health. Some pathogenic microorganisms as well as members of the commensal intestinal microbiota have been shown to be able to break down these secreted compounds. Our understanding of host-compound degradation by the commensal microbiota has been limited to knowledge about simplified model systems because of the difficulty in studying the complex intestinal ecosystem in vivo. In this study, we introduce an approach that overcomes previous technical limitations and allows us to observe which microbial cells in the intestine use host-derived compounds. We added stable isotope-labeled threonine i.v. to mice and combined fluorescence in situ hybridization with high-resolution secondary ion mass spectrometry imaging to characterize utilization of host proteins by individual bacterial cells. We show that two bacterial species, Bacteroides acidifaciens and Akkermansia muciniphila, are important host-protein foragers in vivo. Using gnotobiotic mice we show that microbiota composition determines the magnitude and pattern of foraging by these organisms, demonstrating that a complex microbiota is necessary in order for this niche to be fully exploited. These results underscore the importance of in vivo studies of intestinal microbiota, and the approach presented in this study will be a powerful tool to address many other key questions in animal and human microbiome research.
Dispersal of thermophilic Desulfotomaculum endospores to Baltic Sea sediments over thousands of years
2013 - ISME J., 7: 72-84
Abstract:
Patterns of microbial biogeography result from a combination of dispersal, speciation and extinction, yet individual contributions exerted by each of these mechanisms are difficult to isolate and distinguish. The influx of endospores of strictly thermophilic microorganisms to cold marine sediments offers a natural model for investigating passive dispersal in the ocean. We investigated the activity, diversity and abundance of thermophilic endospore-forming sulfate-reducing bacteria (SRB) in Aarhus Bay by incubating pasteurized sediment between 28 and 85°C, and by subsequent molecular diversity analyses of 16S rRNA and dsrAB genes within the endospore-forming SRB genus Desulfotomaculum, which encompasses the majority of known endospore-forming SRB species. The thermophilic Desulfotomaculum community in Aarhus Bay sediments consisted of at least 23 species level 16S rRNA sequence phylotypes. In two cases, pairs of identical 16S rRNA and dsrAB sequences in Arctic surface sediment 3000 km away showed that the same phylotypes are present in both locations. Radiotracer-enhanced most probable number analysis revealed that the abundance of endospores of thermophilic SRB in Aarhus Bay sediment was ca. 104 cm-3 at the surface and decreased exponentially to 100 cm-3 at 6.5 m depth, corresponding to 4500 years of sediment age. Thus a half-life of ca. 300 years was estimated for the thermophilic SRB endospores deposited in Aarhus Bay sediments. These endospores were similarly detected in the overlying water column, indicative of passive dispersal in water masses preceding sedimentation. The sources of these thermophiles remain enigmatic, but at least one source may be common to both Aarhus Bay and Arctic sediments.
Three manganese oxide-rich marine sediments harbor similar communities of acetate-oxidizing manganese-reducing bacteria
2012 - ISME J., 6: 2078-90
Abstract:
Dissimilatory manganese reduction dominates anaerobic carbon oxidation in marine sediments with high manganese oxide concentrations, but the microorganisms responsible for this process are largely unknown. In this study, the acetate-utilizing manganese-reducing microbiota in geographically well-separated, manganese oxide-rich sediments from Gullmar Fjord (Sweden), Skagerrak (Norway) and Ulleung Basin (Korea) were analyzed by 16S rRNA-stable isotope probing (SIP). Manganese reduction was the prevailing terminal electron-accepting process in anoxic incubations of surface sediments, and even the addition of acetate stimulated neither iron nor sulfate reduction. The three geographically distinct sediments harbored surprisingly similar communities of acetate-utilizing manganese-reducing bacteria: 16S rRNA of members of the genera Colwellia and Arcobacter and of novel genera within the Oceanospirillaceae and Alteromonadales were detected in heavy RNA-SIP fractions from these three sediments. Most probable number (MPN) analysis yielded up to 10(6) acetate-utilizing manganese-reducing cells cm(-3) in Gullmar Fjord sediment. A 16S rRNA gene clone library that was established from the highest MPN dilutions was dominated by sequences of Colwellia and Arcobacter species and members of the Oceanospirillaceae, supporting the obtained RNA-SIP results. In conclusion, these findings strongly suggest that (i) acetate-dependent manganese reduction in manganese oxide-rich sediments is catalyzed by members of taxa (Arcobacter, Colwellia and Oceanospirillaceae) previously not known to possess this physiological function, (ii) similar acetate-utilizing manganese reducers thrive in geographically distinct regions and (iii) the identified manganese reducers differ greatly from the extensively explored iron reducers in marine sediments.
Complete genome sequences of Desulfosporosinus orientis DSM765T, Desulfosporosinus youngiae DSM17734T, Desulfosporosinus meridiei DSM13257T, and Desulfosporosinus acidiphilus DSM22704T
2012 - J. Bacteriol., 194: 6300-1
Abstract:
Desulfosporosinus species are sulfate-reducing bacteria belonging to the Firmicutes. Their genomes will give insights into the genetic repertoire and evolution of sulfate reducers typically thriving in terrestrial environments and able to degrade toluene (Desulfosporosinus youngiae), to reduce Fe(III) (Desulfosporosinus meridiei, Desulfosporosinus orientis), and to grow under acidic conditions (Desulfosporosinus acidiphilus).
Modeling formamide denaturation of probe-target hybrids for improved microarray probe design in microbial diagnostics
2012 - PLoS One, 7: e43862
Abstract:
Application of high-density microarrays to the diagnostic analysis of microbial communities is challenged by the optimization of oligonucleotide probe sensitivity and specificity, as it is generally unfeasible to experimentally test thousands of probes. This study investigated the adjustment of hybridization stringency using formamide with the idea that sensitivity and specificity can be optimized during probe design if the hybridization efficiency of oligonucleotides with target and non-target molecules can be predicted as a function of formamide concentration. Sigmoidal denaturation profiles were obtained using fluorescently labeled and fragmented 16S rRNA gene amplicon of Escherichia coli as the target with increasing concentrations of formamide in the hybridization buffer. A linear free energy model (LFEM) was developed and microarray-specific nearest neighbor rules were derived. The model simulated formamide melting with a denaturant m-value that increased hybridization free energy (ΔG°) by 0.173 kcal/mol per percent of formamide added (v/v). Using the LFEM and specific probe sets, free energy rules were systematically established to predict the stability of single and double mismatches, including bulged and tandem mismatches. The absolute error in predicting the position of experimental denaturation profiles was less than 5% formamide for more than 90 percent of probes, enabling a practical level of accuracy in probe design. The potential of the modeling approach for probe design and optimization is demonstrated using a dataset including the 16S rRNA gene of Rhodobacter sphaeroides as an additional target molecule. The LFEM and thermodynamic databases were incorporated into a computational tool (ProbeMelt) that is freely available at http://DECIPHER.cee.wisc.edu
Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis
2012 - ISME J., 6: 2091-106
Abstract:
Human inflammatory bowel disease and experimental colitis models in mice are associated with shifts in intestinal microbiota composition, but it is unclear at what taxonomic/phylogenetic level such microbiota dynamics can be indicative for health or disease. Here, we report that dextran sodium sulfate (DSS)-induced colitis is accompanied by major shifts in the composition and function of the intestinal microbiota of STAT1(-/-) and wild-type mice, as determined by 454 pyrosequencing of bacterial 16S rRNA (gene) amplicons, metatranscriptomics and quantitative fluorescence in situ hybridization of selected phylotypes. The bacterial families Ruminococcaceae, Bacteroidaceae, Enterobacteriaceae, Deferribacteraceae and Verrucomicrobiaceae increased in relative abundance in DSS-treated mice. Comparative 16S rRNA sequence analysis at maximum possible phylogenetic resolution identified several indicator phylotypes for DSS treatment, including the putative mucin degraders Akkermansia and Mucispirillum. The analysis additionally revealed strongly contrasting abundance changes among phylotypes of the same family, particularly within the Lachnospiraceae. These extensive phylotype-level dynamics were hidden when reads were grouped at higher taxonomic levels. Metatranscriptomic analysis provided insights into functional shifts in the murine intestinal microbiota, with increased transcription of genes associated with regulation and cell signaling, carbohydrate metabolism and respiration and decreased transcription of flagellin genes during inflammation. These findings (i) establish the first in-depth inventory of the mouse gut microbiota and its metatranscriptome in the DSS colitis model, (ii) reveal that family-level microbial community analyses are insufficient to reveal important colitis-associated microbiota shifts and (iii) support a scenario of shifting intra-family structure and function in the phylotype-rich and phylogenetically diverse Lachnospiraceae in DSS-treated mice.
Sulfate-reducing microorganisms in wetlands - fameless actors in carbon cycling and climate change
2012 - Frontiers Microbiol., 3: 72
Abstract:
Freshwater wetlands are a major source of the greenhouse gas methane but at the same time can function as carbon sink. Their response to global warming and environmental pollution is one of the largest unknowns in the upcoming decades to centuries. In this review, we highlight the role of sulfate-reducing microorganisms (SRM) in the intertwined element cycles of wetlands. Although regarded primarily as methanogenic environments, biogeochemical studies have revealed a previously hidden sulfur cycle in wetlands that can sustain rapid renewal of the small standing pools of sulfate. Thus, dissimilatory sulfate reduction, which frequently occurs at rates comparable to marine surface sediments, can contribute up to 36–50% to anaerobic carbon mineralization in these ecosystems. Since sulfate reduction is thermodynamically favored relative to fermentative processes and methanogenesis, it effectively decreases gross methane production thereby mitigating the flux of methane to the atmosphere. However, very little is known about wetland SRM. Molecular analyses using dsrAB [encoding subunit A and B of the dissimilatory (bi)sulfite reductase] as marker genes demonstrated that members of novel phylogenetic lineages, which are unrelated to recognized SRM, dominate dsrAB richness and, if tested, are also abundant among the dsrAB-containing wetland microbiota. These discoveries point toward the existence of so far unknown SRM that are an important part of the autochthonous wetland microbiota. In addition to these numerically dominant microorganisms, a recent stable isotope probing study of SRM in a German peatland indicated that rare biosphere members might be highly active in situ and have a considerable stake in wetland sulfate reduction. The hidden sulfur cycle in wetlands and the fact that wetland SRM are not well represented by described SRM species explains their so far neglected role as important actors in carbon cycling and climate change.
amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions
2012 - Environ. Microbiol., 14: 525-539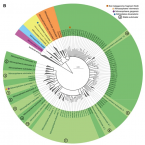
Abstract:
Ammonia-oxidizing archaea (AOA) play an important role in nitrification and many studies exploit their amoA genes as marker for their diversity and abundance. We present an archaeal amoA consensus phylogeny based on all publicly available sequences (status June 2010) and provide evidence for the diversification of AOA into four previously recognized clusters and one newly identified major cluster. These clusters, for which we suggest a new nomenclature, harbored 83 AOA species-level OTU (using an inferred species threshold of 85% amoA identity). 454 pyrosequencing of amoA amplicons from 16 soils sampled in Austria, Costa Rica, Greenland, and Namibia revealed that only 2% of retrieved sequences had no database representative on the species-level and represented 30–37 additional species-level OTUs. With the exception of an acidic soil from which mostly amoA amplicons of the Nitrosotalea cluster were retrieved, all soils were dominated by amoA amplicons from the Nitrososphaera cluster (also called group I.1b), indicating that the previously reported AOA from the Nitrosopumilus cluster (also called group I.1a) are absent or represent minor populations in soils. AOA richness estimates on the species level ranged from 8–83 co-existing AOAs per soil. Presence/absence of amoA OTUs (97% identity level) correlated with geographic location, indicating that besides contemporary environmental conditions also dispersal limitation across different continents and/or historical environmental conditions might influence AOA biogeography in soils.
Barcoded primers used in multiplex amplicon pyrosequencing bias amplification
2011 - Appl. Environ. Microbiol., 77: 7846-7849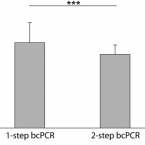
Abstract:
"Barcode-tagged" PCR primers used for multiplex amplicon sequencing generate a thus-far-overlooked amplification bias that produces variable terminal restriction fragment length polymorphism (T-RFLP) and pyrosequencing data from the same environmental DNA template. We propose a simple two-step PCR approach that increases reproducibility and consistently recovers higher genetic diversity in pyrosequencing libraries.
Systematic spatial bias in DNA microarray hybridization is caused by probe spot position-dependent variability in lateral diffusion
2011 - PLoS One, 6: e23727
Abstract:
The hybridization of nucleic acid targets with surface-immobilized probes is a widely used assay for the parallel detection of multiple targets in medical and biological research. Despite its widespread application, DNA microarray technology still suffers from several biases and lack of reproducibility, stemming in part from an incomplete understanding of the processes governing surface hybridization. In particular, non-random spatial variations within individual microarray hybridizations are often observed, but the mechanisms underpinning this positional bias remain incompletely explained.
METHODOLOGY/PRINCIPAL FINDINGS:
This study identifies and rationalizes a systematic spatial bias in the intensity of surface hybridization, characterized by markedly increased signal intensity of spots located at the boundaries of the spotted areas of the microarray slide. Combining observations from a simplified single-probe block array format with predictions from a mathematical model, the mechanism responsible for this biasis found to be a position-dependent variation in lateral diffusion of target molecules. Numerical simulations reveal a strong influence of microarraywell geometry on the spatial bias.
CONCLUSIONS:
Reciprocal adjustment of the size of the microarray hybridization chamber to the area of surface-bound probes is a simple and effective measure to minimize or eliminate the diffusion-based bias, resulting in increased uniformity and accuracy of quantitative DNA microarrayhybridization.
Paracatenula, an ancient symbiosis between thiotrophic Alphaproteobacteria and catenulid flatworms
2011 - Proc. Natl. Acad. Sci. USA, 108: 12078-12083
Abstract:
Harnessing chemosynthetic symbionts is a recurring evolutionary strategy. Eukaryotes from six phyla as well as one archaeon have acquired chemoautotrophic sulfur-oxidizing bacteria. In contrast to this broad host diversity, known bacterial partners apparently belong to two classes of bacteria-the Gamma- and Epsilonproteobacteria. Here, we characterize the intracellular endosymbionts of the mouthless catenulid flatworm genus Paracatenula as chemoautotrophic sulfur-oxidizing Alphaproteobacteria. The symbionts of Paracatenula galateia are provisionally classified as "Candidatus Riegeria galateiae" based on 16S ribosomal RNA sequencing confirmed by fluorescence in situ hybridization together with functional gene and sulfur metabolite evidence. 16S rRNA gene phylogenetic analysis shows that all 16 Paracatenula species examined harbor host species-specific intracellular Candidatus Riegeria bacteria that form a monophyletic group within the order Rhodospirillales. Comparing host and symbiont phylogenies reveals strict cocladogenesis and points to vertical transmission of the symbionts. Between 33% and 50% of the body volume of the various worm species is composed of bacterial symbionts, by far the highest proportion among all known endosymbiotic associations between bacteria and metazoans. This symbiosis, which likely originated more than 500 Mya during the early evolution of flatworms, is the oldest known animal-chemoautotrophic bacteria association. The distant phylogenetic position of the symbionts compared with other mutualistic or parasitic Alphaproteobacteria promises to illuminate the common genetic predispositions that have allowed several members of this class to successfully colonize eukaryote cells.
Microorganisms with novel dissimilatory (bi)sulfite reductase genes are widespread and part of the core microbiota in low-sulfate peatlands
2011 - Appl. Environ. Microbiol., 77: 1231-1242
Abstract:
Peatlands of the Lehstenbach catchment (Germany) house as-yet-unidentified microorganisms with phylogenetically novel variants of the dissimilatory (bi)sulfite reductase genes dsrAB. These genes are characteristic of microorganisms that reduce sulfate, sulfite, or some organosulfonates for energy conservation but can also be present in anaerobic syntrophs. However, nothing is currently known regarding the abundance, community dynamics, and biogeography of these dsrAB-carrying microorganisms in peatlands. To tackle these issues, soils from a Lehstenbach catchment site (Schlöppnerbrunnen II fen) from different depths were sampled at three time points over a 6-year period to analyze the diversity and distribution of dsrAB-containing microorganisms by a newly developed functional gene microarray and quantitative PCR assays. Members of novel, uncultivated dsrAB lineages (approximately representing species-level groups) (i) dominated a temporally stable but spatially structured dsrAB community and (ii) represented "core" members (up to 1% to 1.7% relative abundance) of the autochthonous microbial community in this fen. In addition, denaturing gradient gel electrophoresis (DGGE)- and clone library-based comparisons of the dsrAB diversity in soils from a wet meadow, three bogs, and five fens of various geographic locations (distance of 1 to 400 km) identified that one Syntrophobacter-related and nine novel dsrAB lineages are widespread in low-sulfate peatlands. Signatures of biogeography in dsrB-based DGGE data were not correlated with geographic distance but could be explained largely by soil pH and wetland type, implying that the distribution of dsrAB-carrying microorganisms in wetlands on the scale of a few hundred kilometers is not limited by dispersal but determined by local environmental conditions.
A 'rare biosphere' microorganism contributes to sulfate reduction in a peatland
2010 - ISME J., 12: 1591-1602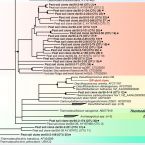
Abstract:
Methane emission from peatlands contributes substantially to global warming but is significantly reduced by sulfate reduction, which is fuelled by globally increasing aerial sulfur pollution. However, the biology behind sulfate reduction in terrestrial ecosystems is not well understood and the key players for this process as well as their abundance remained unidentified. Comparative 16S rRNA gene stable isotope probing (SIP) in the presence and absence of sulfate indicated that a Desulfosporosinus species, which constitutes only 0.006% of the total microbial community 16S rRNA genes, is an important sulfate reducer in a long-term experimental peatland field site. Parallel SIP using dsrAB (encoding subunit A and B of the dissimilatory (bi)sulfite reductase) identified no additional sulfate reducers under the conditions tested. For the identified Desulfosporosinus species a high cell-specific sulfate reduction rate of up to 341 fmol SO(4)(2-) cell(-1) day(-1) was estimated. Thus, the small Desulfosporosinus population has the potential to reduce sulfate in situ at a rate of 4.0-36.8 nmol (g soil w. wt.)(-1) day(-1), sufficient to account for a considerable part of sulfate reduction in the peat soil. Modeling of sulfate diffusion to such highly active cells identified no limitation in sulfate supply even at bulk concentrations as low as 10 muM. Collectively, these data show that the identified Desulfosporosinus species, despite being a member of the 'rare biosphere', contributes to an important biogeochemical process that diverts the carbon flow in peatlands from methane to CO(2) and, thus, alters their contribution to global warming.
Thermophilic anaerobes in Arctic marine sediments induced to mineralize complex organic matter at high temperature
2010 - Environ. Microbiol., 12: 1089-1104Abstract:
Summary Marine sediments harbour diverse populations of dormant thermophilic bacterial spores that become active in sediment incubation experiments at much higher than in situ temperature. This response was investigated in the presence of natural complex organic matter in sediments of two Arctic fjords, as well as with the addition of freeze-dried Spirulina or individual high-molecular-weight polysaccharides. During 50 degrees C incubation experiments, Arctic thermophiles catalysed extensive mineralization of the organic matter via extracellular enzymatic hydrolysis, fermentation and sulfate reduction. This high temperature-induced food chain mirrors sediment microbial processes occurring at cold in situ temperatures (near 0 degrees C), yet it is catalysed by a completely different set of microorganisms. Using sulfate reduction rates (SRR) as a proxy for organic matter mineralization showed that differences in organic matter reactivity determined the extent of the thermophilic response. Fjord sediments with higher in situ SRR also supported higher SRR at 50 degrees C. Amendment with Spirulina significantly increased volatile fatty acids production and SRR relative to unamended sediment in 50 degrees C incubations. Spirulina amendment also revealed temporally distinct sulfate reduction phases, consistent with 16S rRNA clone library detection of multiple thermophilic Desulfotomaculum spp. enriched at 50 degrees C. Incubations with four different fluorescently labelled polysaccharides at 4 degrees C and 50 degrees C showed that the thermophilic population in Arctic sediments produce a different suite of polymer-hydrolysing enzymes than those used in situ by the cold-adapted microbial community. Over time, dormant marine microorganisms like these are buried in marine sediments and might eventually encounter warmer conditions that favour their activation. Distinct enzymatic capacities for organic polymer degradation could allow specific heterotrophic populations like these to play a role in sustaining microbial metabolism in the deep, warm, marine biosphere.
A constant flux of diverse thermophilic bacteria into the cold Arctic seabed
2009 - Science, 325: 1541-1544Abstract:
Microorganisms have been repeatedly discovered in environments that do not support their metabolic activity. Identifying and quantifying these misplaced organisms can reveal dispersal mechanisms that shape natural microbial diversity. Using endospore germination experiments, we estimated a stable supply of thermophilic bacteria into permanently cold Arctic marine sediment at a rate exceeding 10(8) spores per square meter per year. These metabolically and phylogenetically diverse Firmicutes show no detectable activity at cold in situ temperatures but rapidly mineralize organic matter by hydrolysis, fermentation, and sulfate reduction upon induction at 50 degrees C. The closest relatives to these bacteria come from warm subsurface petroleum reservoir and ocean crust ecosystems, suggesting that seabed fluid flow from these environments is delivering thermophiles to the cold ocean. These transport pathways may broadly influence microbial community composition in the marine environment.
Isotope array analysis of Rhodocyclales uncovers functional redundancy and versatility in an activated sludge
2009 - ISME J., 3: 1349-1364
Abstract:
Extensive physiological analyses of different microbial community members in many samples are difficult because of the restricted number of target populations that can be investigated in reasonable time by standard substrate-mediated isotope-labeling techniques. The diversity and ecophysiology of Rhodocyclales in activated sludge from a full-scale wastewater treatment plant were analyzed following a holistic strategy based on the isotope array approach, which allows for a parallel functional probing of different phylogenetic groups. Initial diagnostic microarray, comparative 16S rRNA gene sequence, and quantitative fluorescence in situ hybridization surveys indicated the presence of a diverse community, consisting of an estimated number of 27 operational taxonomic units that grouped in at least seven main Rhodocyclales lineages. Substrate utilization profiles of probe-defined populations were determined by radioactive isotope array analysis and microautoradiography-fluorescence in situ hybridization of activated sludge samples that were briefly exposed to different substrates under oxic and anoxic, nitrate-reducing conditions. Most detected Rhodocyclales groups were actively involved in nitrogen transformation, but varied in their consumption of propionate, butyrate, or toluene, and thus in their ability to use different carbon sources in activated sludge. This indicates that the functional redundancy of nitrate reduction and the functional versatility of substrate usage are important factors governing niche overlap and differentiation of diverse Rhodocyclales members in this activated sludge.
16S rRNA gene-based phylogenetic microarray for simultaneous identification of members of the genus Burkholderia
2009 - Environ. Microbiol., 11: 779-800Abstract:
For cultivation-independent and highly parallel analysis of members of the genus Burkholderia, an oligonucleotide microarray (phylochip) consisting of 131 hierarchically nested 16S rRNA gene-targeted oligonucleotide probes was developed. A novel primer pair was designed for selective amplification of a 1.3 kb 16S rRNA gene fragment of Burkholderia species prior to microarray analysis. The diagnostic performance of the microarray for identification and differentiation of Burkholderia species was tested with 44 reference strains of the genera Burkholderia, Pandoraea, Ralstonia and Limnobacter. Hybridization patterns based on presence/absence of probe signals were interpreted semi-automatically using the novel likelihood-based strategy of the web-tool Phylo- Detect. Eighty-eight per cent of the reference strains were correctly identified at the species level. The evaluated microarray was applied to investigate shifts in the Burkholderia community structure in acidic forest soil upon addition of cadmium, a condition that selected for Burkholderia species. The microarray results were in agreement with those obtained from phylogenetic analysis of Burkholderia 16S rRNA gene sequences recovered from the same cadmiumcontaminated soil, demonstrating the value of the Burkholderia phylochip for determinative and environmental studies.
High genetic similarity between two geographically distinct strains of the sulfur-oxidizing symbiont 'Candidatus Thiobios zoothamnicoli'
2009 - FEMS Microbiol. Ecol., 67: 229-41
Abstract:
The giant marine ciliate Zoothamnium niveum (Ciliophora, Oligohymenophora) is obligatorily covered by a monolayer of putative chemoautotrophicsulfur-oxidizing (thiotrophic) bacteria. For Z. niveum specimens from the Caribbean Sea it has been demonstrated that this ectosymbiotic population consists of only a single pleomorphic phylotype described as Candidatus Thiobios zoothamnicoli. The goal of our study was to identify and phylogenetically analyse the ectosymbiont(s) of a recently discovered Z. niveum population from the Mediterranean Sea, and to compare marker genes encoding key enzymes of the carbon and sulfur metabolism between the two symbiont populations. We identified a single bacterial phylotype representing the ectosymbiont of Z. niveum from the Mediterranean population showing 99.7% 16S rRNA gene (99.2% intergenic spacer region)similarity to the Caribbean Z. niveum ectosymbiont. Genes encoding enzymes typical for an inorganic carbon metabolism [ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO)] and for sulfur metabolism (5'-adenylylsulfate reductase, dissimilatory sulfite reductase) were detected in both symbiotic populations. The very high amino acid sequence identity (97-100%) and the high nucleic acid sequence identity (90-98%) of these marker enzymes in two geographically distant symbiont populations suggests that the association of Z. niveum with Cand. Thiobios zoothamnicoli is very specific as well as temporally and spatially stable.
Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes
2009 - Environ. Microbiol., 11: 289-299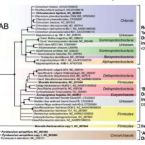
Abstract:
Sulfur-oxidizing prokaryotes (SOP) catalyse a central step in the global S-cycle and are of major functional importance for a variety of natural and engineered systems, but our knowledge on their actual diversity and environmental distribution patterns is still rather limited. In this study we developed a specific PCR assay for the detection of dsrAB that encode the reversely operating sirohaem dissimilatory sulfite reductase (rDSR) and are present in many but not all published genomes of SOP. The PCR assay was used to screen 42 strains of SOP (most without published genome sequence) representing the recognized diversity of this guild. For 13 of these strains dsrAB was detected and the respective PCR product was sequenced. Interestingly, most dsrAB-encoding SOP are capable of forming sulfur storage compounds. Phylogenetic analysis demonstrated largely congruent rDSR and 16S rRNA consensus tree topologies, indicating that lateral transfer events did not play an important role in the evolutionary history of known rDSR. Thus, this enzyme represents a suitable phylogenetic marker for diversity analyses of sulfur storage compound-exploiting SOP in the environment. The potential of this new functional gene approach was demonstrated by comparative sequence analyses of all dsrAB present in published metagenomes and by applying it for a SOP census in selected marine worms and an alkaline lake sediment.
Multiple bacterial symbionts in two species of co-occurring gutless oligochaete worms from Mediterranean sea grass sediments
2008 - Environ Microbiol., 10: 3404-3416Abstract:
Gutless oligochaete worms are found worldwide in the pore waters of marine sediments and live in symbiosis with chemoautotrophic sulfur-oxidizingbacteria. In the Mediterranean, two species of gutless oligochaete worms, Olavius algarvensis and O. ilvae, co-occur in sediments around sea grass beds. These sediments have extremely low sulfide concentrations (< 1 microM), raising the question if O. ilvae, as shown previously for O. algarvensis, also harbours sulfate-reducing symbionts that provide its sulfur-oxidizing symbionts with reduced sulfur compounds. In this study, we used fluorescence in situ hybridization (FISH) and comparative sequence analysis of genes for 16S rRNA, sulfur metabolism (aprA and dsrAB), and autotrophic carbon fixation (cbbL) to examine the microbial community of O. ilvae and re-examine the O. algarvensis symbiosis. In addition to the four previously described symbionts of O. algarvensis, in this study a fifth symbiont belonging to the Spirochaetes was found in these hosts. The symbiotic community of O. ilvae was similar to that of O. algarvensis and also included two gammaproteobacterial sulfur oxidizers and two deltaproteobacterial sulfate reducers, but not a spirochete. The phylogenetic and metabolic similarity of the symbiotic communities in these two co-occurring host species that are not closely related to each other indicates that syntrophic sulfur cycling provides a strong selective advantage to these worms in their sulfide-poor environment.
probeCheck - a central resource for evaluating oligonucleotide probe coverage and specificity
2008 - Environ. Microbiol., 10: 2894-2896
Abstract:
The web server probeCheck, freely accessible at www.microbial-ecology.net/probecheck, provides a pivotal forum for rapid specificity and coverage evaluations of probes and primers against selected databases of phylogenetic and functional marker genes. Currently, 24 widely used sequence collections including the Ribosomal Database Project (RDP) II, Greengenes, SILVA, and the Functional Gene Pipeline/Repository can be queried. For this purpose, probeCheck integrates a new online version of the popular ARB probe match tool with free energy (ΔG) calculations for each perfectly-matched and mismatched probe-target hybrid, allowing assessment of the theoretical binding stabilities of oligo-target and non-target hybrids. For each output sequence, the accession number, the GenBank taxonomy, and a link to the respective entry at GenBank, EMBL, and, if applicable, the query database is displayed. Filtering options allow customizing results on the output page. In addition, probeCheck is linked with probe match tools of RDP II and Greengenes, NCBI Blast, the Oligonucleotide Properties Calculator, the two-state folding tool of the DINAMelt server, and the rRNA-targeted probe database probeBase. Taken together, these features provide a multifunctional platform with maximal flexibility for the user in the choice of databases and options for the evaluation of published and newly developed probes and primers.
Biogeography of sulfate-reducing prokaryotes in river floodplains
2008 - FEMS Microbiol. Ecol., 64: 395-406Abstract:
In this study, a large-scale field survey was conducted to describe the biogeography of sulfate-reducing prokaryotes (SRPs) in river floodplains. Fingerprints obtained with three methods, i.e. 16S rRNA gene-based oligonucleotide microarray, dsrB-based denaturing gradient gel electrophoresis (DGGE) and polar lipid-derived fatty acid (PLFA) analyses, were used as a proxy to describe the SRPs community diversity. Each set of profiles was subjected to a combined multivariate/correlation analysis in order to compare SRP community profiles and to highlight the environmental variables influencing the SRPs distribution along environmental gradients. Floodplain soils harbored distinct SRP communities displaying biogeographic patterns. Nearly all profiles from the tidal sites consistently separated from the nontidal sites, independently from the screening method and the multivariate statistics used. The distribution of the microarray/DGGE/PLFA-based fingerprints in the principal component plots could be correlated to eight soil variables, i.e. soil organic matter, total nitrogen, total phosphorous and total potassium, and extractable ammonium, nitrate, phosphate and sulfate, as well as seven pore water variables, i.e. phosphate, sulfate, sulfide, chloride, sodium, potassium and magnesium ions. Indication of a salinity- and plant nutrient-dependent distribution of SRPs related to Desulfosarcina, Desulfomonile and Desulfobacter was suggested by microarray, DGGE and PLFA analyses.
Improved 16S rRNA-targeted oligonucleotide probe set for analysis of sulfate-reducing bacteria by fluorescence in situ hybridization
2007 - J. Microbiol. Methods, 69: 523-528Abstract:
An updated dataset of in silico specificities for 54 previously published 16S rRNA-targeted oligonucleotides was assembled to provide guidance for reliable fluorescence in situ hybridization (FISH) analysis of sulfate-reducing bacteria. Additionally, six new FISH probes were developed for major deltaproteobacterial taxa, including a probe trio targeting most Deltaproteobacteria and Gemmatimonadetes.
Diversity of sulfate-reducing bacteria from an extreme hypersaline sediment in Great Salt Lake (Utah, USA)
2007 - FEMS Microbiol. Ecol., 60: 287-298
Abstract:
The diversity of sulfate-reducing bacteria (SRB) inhabiting the extreme hypersaline sediment (270 g L(-1) NaCl) of the northern arm of Great Salt Lake was studied by integrating cultivation and genotypic identification approaches involving PCR-based retrieval of 16S rRNA and dsrAB genes, the latter encoding major subunits of dissimilatory (bi) sulfite reductase. The majority (85%) of dsrAB sequences retrieved directly from the sediment formed a lineage of high (micro) diversity affiliated with the genus Desulfohalobium, while others represented novel lineages within the families Desulfohalobiaceae and Desulfobacteraceae or among Gram-positive SRB. Using the same sediment, SRB enrichment cultures were established in parallel at 100 and at 190 g L(-1) NaCl using different electron donors. After 5-6 transfers, dsrAB and 16S rRNA gene-based profiling of these enrichment cultures recovered a SRB community composition congruent with the cultivation-independent profiling of the sediment. Pure culture representatives of the predominant Desulfohalobium-related lineage and of one of the Desulfobacteraceae-affilated lineages were successfully obtained. The growth performance of these isolates and of the enrichment cultures suggests that the sediment SRB community of the northern arm of Great Salt Lake consists of moderate halophiles, which are salt-stressed at the in situ salinity of 27%.
Unravelling microbial communities with DNA-microarrays: challenges and future directions
2007 - Microb. Ecol., 53: 498-506
Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea
2007 - Environ. Microbiol., 9: 131-142
Abstract:
The Black Sea, with its highly sulfidic water column, is the largest anoxic basin in the world. Within its sediments, the mineralization of organic matter occurs essentially through sulfate reduction and methanogenesis. In this study, the sulfate-reducing community was investigated in order to understand how these microorganisms are distributed relative to the chemical zonation: in the upper sulfate zone, at the sulfate-methane transition zone, and deeply within the methane zone. Total bacteria were quantified by real-time PCR of 16S rRNA genes whereas sulfate-reducing microorganisms (SRM) were quantified by targeting their metabolic key gene, the dissimilatory (bi)sulfite reductase (dsrA). Sulfate-reducing microorganisms were predominant in the sulfate zone but occurred also in the methane zone, relative proportion was maximal around the sulfate-methane transition, c. 30%, and equally high in the sulfate and methane zones, 5-10%. The dsrAB clone library from the sulfate-methane transition zone, showed mostly sequences affiliated with the Desulfobacteraceae. While, the dsrAB clone libraries from the upper, sulfate-rich zone and the deep, sulfate-poor zone were dominated by similar, novel deeply branching sequences which might represent Gram-positive spore-forming sulfate- and/or sulfite-reducing microorganisms. We thus hypothesize that terminal carbon mineralization in surface sediments of the Black Sea is largely due to the sulfate reduction activity of previously hidden SRM. Although these novel SRM were also abundant in sulfate-poor, methanogenic areas of the Black Sea sediment, their activities and possibly very versatile metabolic capabilities remain subject of further study.
probeBase - an online resource for rRNA-targeted oligonucleotide probes: new features 2007
2007 - Nucleic Acids Res., 35: D800-D804
Abstract:
probeBase is a curated database of annotated rRNA-targeted oligonucleotide probes and supporting information. Rapid access to probe, microarray and reference data is achieved by powerful search tools and via different lists that are based on selected categories such as functional or taxonomic properties of the target organism(s), or the hybridization format (fluorescence in situ hybridization or microarray) in which the probes were applied. Additional information on probe coverage and specificity is available through direct submissions of probe sequences from probeBase to RDP-II and Greengenes, two major rRNA sequence databases. A freely-editable user comments field for each probe entry allows any user to add, modify, or remove information, or to report errors in real-time. probeBase entries increased from 700 to more than 1,200 during the past three years. Several options for submission of single probes or entire probe sets, even prior to publication of newly developed probes, should further contribute to keeping probeBase an up-to-date and useful resource. probeBase is freely accessible at http://www.microbial-ecology.net/probebase. Email correspondence can be addressed to probebase.dome@univie.ac.at.
Non-sulfate-reducing, syntrophic bacteria affiliated with the Desulfotomaculum cluster I are widely distributed in methanogenic environments
2006 - Appl. Environ. Microbiol., 72: 2080-2091
Abstract:
The classical perception of members of the gram-positive Desulfotomaculum cluster I as sulfate-reducing bacteria was recently challenged by the isolation of new representatives lacking the ability for anaerobic sulfate respiration. For example, the two described syntrophic propionate-oxidizing species of the genus Pelotomaculum form the novel Desulfotomaculum subcluster Ih. In the present study, we applied a polyphasic approach by using cultivation-independent and culturing techniques in order to further characterize the occurrence, abundance, and physiological properties of subcluster Ih bacteria in low-sulfate, methanogenic environments. 16S rRNA (gene)-based cloning, quantitative fluorescence in situ hybridization, and real-time PCR analyses showed that the subcluster Ih population composed a considerable part of the Desulfotomaculum cluster I community in almost all samples examined. Additionally, five propionate-degrading syntrophic enrichments of subcluster Ih bacteria were successfully established, from one of which the new strain MGP was isolated in coculture with a hydrogenotrophic methanogen. None of the cultures analyzed, including previously described Pelotomaculum species and strain MGP, consumed sulfite, sulfate, or organosulfonates. In accordance with these phenotypic observations, a PCR-based screening for dsrAB (key genes of the sulfate respiration pathway encoding the alpha and beta subunits of the dissimilatory sulfite reductase) of all enrichments/(co)cultures was negative with one exception. Surprisingly, strain MGP contained dsrAB, which were transcribed in the presence and absence of sulfate. Based on these and previous findings, we hypothesize that members of Desulfotomaculum subcluster Ih have recently adopted a syntrophic lifestyle to thrive in low-sulfate, methanogenic environments and thus have lost their ancestral ability for dissimilatory sulfate/sulfite reduction.
Linking microbial community structure with function: fluorescence in situ hybridization-microautoradiography and isotope arrays
2006 - Curr. Opin. Biotechnol., 17: 1-9
Abstract:
The ecophysiology of microorganisms has been at the heart of microbial ecology since its early days, but only during the past decade have methods become available for cultivation-independent, direct identification of microorganisms in complex communities and for the simultaneous investigation of their activity and substrate uptake patterns. The combination of fluorescence in situ hybridization (FISH) and microautoradiography (MAR) is currently the most widely applied tool for revealing physiological properties of microorganisms in their natural environment with single-cell resolution. For example, this technique has been used in wastewater treatment and marine systems to describe the functional properties of newly discovered species, and to identify microorganisms responsible for key physiological processes. Recently, the scope of FISH-MAR was extended by rendering it quantitative and by combining it with microelectrode measurements or stable isotope probing. Isotope arrays have also been developed that exploit the parallel detection offered by DNA microarrays to measure incorporation of labelled substrate into the rRNA of many community members in a single experiment.
Highly parallel microbial diagnostics using oligonucleotide microarrays
2006 - Clinica Chimica Acta, 363: 106-119Abstract:
Oligonucleotide microarrays are highly parallel hybridization platforms, allowing rapid and simultaneous identification of many different microorganisms and viruses in a single assay. In the past few years, researchers have been confronted with a dramatic increase in the number of studies reporting development and/or improvement of oligonucleotide microarrays for microbial diagnostics, but use of the technology in routine diagnostics is still constrained by a variety of factors. Careful development of microarray essentials (such as oligonucleotide probes, protocols for target preparation and hybridization, etc.) combined with extensive performance testing are thus mandatory requirements for the maturation of diagnostic microarrays from fancy technological gimmicks to robust and routinely applicable tools.
Functional marker genes for identification of sulphate-reducing prokaryotes
2005 - Methods Enzymol., 397: 469-489Abstract:
Sulfate-reducing prokaryotes (SRPs) exploit sulfate as an electron acceptor for anaerobic respiration and exclusively catalyze this essential step of the world's sulfur cycle. Because SRPs are found in many prokaryotic phyla and are often closely related to non-SRPs, 16S rRNA gene-based analyses are inadequate to identify novel lineages of this guild in a cultivation-independent manner. This problem can be solved by comparative sequence analysis of environmentally retrieved gene fragments of the dissimilatory (bi)sulfite (dsrAB) and adenosine-5'-phosphosulfate reductases (apsA), which encode key enzymes of the SRP energy metabolism. This chapter provides detailed protocols for the application of these functional marker molecules for SRP diversity surveys in the environment. Data from the analysis of dsrAB sequence diversity in water samples from the Mariager Fjord in northeast Denmark are presented to illustrate the different steps of the protocols. Furthermore, this chapter describes a novel gel retardation-based technique, suitable for fingerprinting of the approximately 1.9-kb-large dsrAB polymerase chain reaction amplification products, which efficiently increases the chance of retrieving rare and novel dsrAB sequence types from environmental samples.
New insights into metabolic properties of marine bacteria encoding proteorhodopsins
2005 - PLoS Biol., 3: e273
Abstract:
Proteorhodopsin phototrophy was recently discovered in oceanic surface waters. In an effort to characterize uncultured proteorhodopsin-exploiting bacteria, large-insert bacterial artificial chromosome (BAC) libraries from the Mediterranean Sea and Red Sea were analyzed. Fifty-five BACs carried diverse proteorhodopsin genes, and we confirmed the function of five. We calculate that proteorhodopsin-exploiting bacteria account for 13% of microorganisms in the photic zone. We further show that some proteorhodopsin-containing bacteria possess a retinal biosynthetic pathway and a reverse sulfite reductase operon, employed by prokaryotes oxidizing sulfur compounds. Thus, these novel phototrophs are an unexpectedly large and metabolically diverse component of the marine microbial surface water.
Diversity of bacteria growing in a natural mineral water after bottling
2005 - Appl. Environ. Microbiol., 71: 3624-3632Abstract:
Bacterial growth occurs in noncarbonated natural mineral waters a few days after filling and storage at room temperature, a phenomenon known for more than 40 years. Using the full-cycle rRNA approach, we monitored the development of the planktonic bacterial community in a noncarbonated natural mineral water after bottling. Seven 16S rRNA gene libraries, comprising 108 clones in total, were constructed from water samples taken at various days after bottling and from two different bottle sizes. Sequence analyses identified 11 operational taxonomic units (OTUs), all but one affiliated with the betaproteobacterial order Burkholderiales (6 OTUs) or the class Alphaproteobacteria (4 OTUs). Fluorescence in situ hybridization (FISH) was applied in combination with DAPI (4',6'-diamidino-2-phenylindole) staining, viability staining, and microscopic counting to quantitatively monitor changes in bacterial community composition. A growth curve similar to that of a bacterium grown in a batch culture was recorded. In contrast to the current perception that Gammaproteobacteria are the most important bacterial components of natural mineral water in bottles, Betaproteobacteria dominated the growing bacterial community and accounted for 80 to 98% of all bacteria detected by FISH in the late-exponential and stationary-growth phases. Using previously published and newly designed genus-specific probes, members of the betaproteobacterial genera Hydrogenophaga, Aquabacterium, and Polaromonas were found to constitute a significant proportion of the bacterial flora (21 to 86% of all bacteria detected by FISH). For the first time, key genera responsible for bacterial growth in a natural mineral water were identified by applying molecular cultivation-independent techniques.
Oligonucleotide microarray for identification of Enterococcus species
2005 - FEMS Microbiol. Lett., 246: 133-142
Abstract:
For detection of most members of the Enterococcaceae, the specificity of a novel oligonucleotide microarray (ECC-PhyloChip) consisting of 41 hierarchically nested 16S or 23S rRNA gene-targeted probes was evaluated with 23 pure cultures (including 19 Enterococcus species). Target nucleic acids were prepared by PCR amplification of a 4.5-kb DNA fragment containing large parts of the 16S and 23S rRNA genes and were subsequently labeled fluorescently by random priming. Each tested member of the Enterococcaceae was correctly identified on the basis of its unique microarray hybridization pattern. The evaluated ECC-PhyloChip was successfully applied for identification of Enterococcus faecium and Enterococcus faecalis in artificially contaminated milk samples demonstrating the utility of the ECC-PhyloChip for parallel identification and differentiation of Enterococcus species in food samples.
16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order Rhodocyclales
2005 - Appl. Environ. Microbiol., 71: 1373-1386
Abstract:
For simultaneous identification of members of the betaproteobacterial order Rhodocyclales in environmental samples, a 16S rRNA gene-targeted oligonucleotide microarray (RHC-PhyloChip) consisting of 79 probes was developed. Probe design was based on phylogenetic analysis of available 16S rRNA sequences from all cultured and as yet uncultured members of the Rhodocyclales. The multiple nested probe set was evaluated for microarray hybridization with 16S rRNA gene PCR amplicons from 29 reference organisms. Subsequently, the RHC-PhyloChip was successfully used for cultivation-independent Rhodocyclales diversity analysis in activated sludge from an industrial wastewater treatment plant. The implementation of a newly designed Rhodocyclales-selective PCR amplification system prior to microarray hybridization greatly enhanced the sensitivity of the RHC-PhyloChip and thus enabled the detection of Rhodocyclales populations with relative abundances of less than 1% of all bacteria (as determined by fluorescence in situ hybridization) in the activated sludge. The presence of as yet uncultured Zoogloea-, Ferribacterium/Dechloromonas-, and Sterolibacterium-related bacteria in the industrial activated sludge, as indicated by the RHC-PhyloChip analysis, was confirmed by retrieval of their 16S rRNA gene sequences and subsequent phylogenetic analysis, demonstrating the suitability of the RHC-PhyloChip as a novel monitoring tool for environmental microbiology.
Lateral gene transfer of dissimilatory (bi)sulfite reductase revisited
2005 - J. Bacteriol., 187: 2203-2208
Microarray and functional gene analyses of sulfate-reducing prokaryotes in low-sulfate, acidic fens reveal cooccurrence of recognized genera and novel lineages.
2004 - Appl. Environ. Microbiol., 70: 6998-7009
Abstract:
Low-sulfate, acidic (approximately pH 4) fens in the Lehstenbach catchment in the Fichtelgebirge mountains in Germany are unusual habitats for sulfate-reducing prokaryotes (SRPs) that have been postulated to facilitate the retention of sulfur and protons in these ecosystems. Despite the low in situ availability of sulfate (concentration in the soil solution, 20 to 200 microM) and the acidic conditions (soil and soil solution pHs, approximately 4 and 5, respectively), the upper peat layers of the soils from two fens (Schlöppnerbrunnen I and II) of this catchment displayed significant sulfate-reducing capacities. 16S rRNA gene-based oligonucleotide microarray analyses revealed stable diversity patterns for recognized SRPs in the upper 30 cm of both fens. Members of the family "Syntrophobacteraceae" were detected in both fens, while signals specific for the genus Desulfomonile were observed only in soils from Schlöppnerbrunnen I. These results were confirmed and extended by comparative analyses of environmentally retrieved 16S rRNA and dissimilatory (bi)sulfite reductase (dsrAB) gene sequences; dsrAB sequences from Desulfobacca-like SRPs, which were not identified by microarray analysis, were obtained from both fens. Hypotheses concerning the ecophysiological role of these three SRP groups in the fens were formulated based on the known physiological properties of their cultured relatives. In addition to these recognized SRP lineages, six novel dsrAB types that were phylogenetically unrelated to all known SRPs were detected in the fens. These dsrAB sequences had no features indicative of pseudogenes and likely represent novel, deeply branching, sulfate- or sulfite-reducing prokaryotes that are specialized colonists of low-sulfate habitats.
probeBase - an online resource for rRNA-targeted oligonucleotide probes
2003 - Nucleic Acids Res., 31: 514-516
Abstract:
Ribosomal RNA-(rRNA)-targeted oligonucleotide probes are widely used for culture-independent identification of microorganisms in environmental and clinical samples. ProbeBase is a comprehensive database containing more than 700 published rRNA-targeted oligonucleotide probe sequences (status August 2002) with supporting bibliographic and biological annotation that can be accessed through the internet at http://www.probebase.net. Each oligonucleotide probe entry contains information on target organisms, target molecule (small- or large-subunit rRNA) and position, G+C content, predicted melting temperature, molecular weight, necessity of competitor probes, and the reference that originally described the oligonucleotide probe, including a link to the respective abstract at PubMed. In addition, probes successfully used for fluorescence in situ hybridization (FISH) are highlighted and the recommended hybridization conditions are listed. ProbeBase also offers difference alignments for 16S rRNA-targeted probes by using the probe match tool of the ARB software and the latest small-subunit rRNA ARB database (release June 2002). The option to directly submit probe sequences to the probe match tool of the Ribosomal Database Project II (RDP-II) further allows one to extract supplementary information on probe specificities. The two main features of probeBase, 'search probeBase' and 'find probe set', help researchers to find suitable, published oligonucleotide probes for microorganisms of interest or for rRNA gene sequences submitted by the user. Furthermore, the 'search target site' option provides guidance for the development of new FISH probes.
Microbial community composition and function in wastewater treatment plants
2002 - Antonie van Leeuwenhoek, 81: 665-680
Abstract:
Biological wastewater treatment has been applied for more than a century to ameliorate anthropogenic damage to the environment. But only during the last decade the use of molecular tools allowed to accurately determine the composition, and dynamics of activated sludge and biofilm microbial communities. Novel, in many cases yet not cultured bacteria were identified to be responsible for filamentous bulking and foaming as well as phosphorus and nitrogen removal in these systems. Now, methods are developed to infer the in situ physiology of these bacteria. Here we provide an overview of what is currently known about the identity and physiology of some of the microbial key players in activated sludge and biofilm systems.
Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment
2002 - Appl. Environ. Microbiol., 68: 5064-5081
Abstract:
For cultivation-independent detection of sulfate-reducing prokaryotes (SRPs) an oligonucleotide microarray consisting of 132 16S rRNA gene-targeted oligonucleotide probes (18-mers) having hierarchical and parallel (identical) specificity for the detection of all known lineages of sulfate-reducing prokaryotes (SRP-PhyloChip) was designed and subsequently evaluated with 41 suitable pure cultures of SRPs. The applicability of SRP-PhyloChip for diversity screening of SRPs in environmental and clinical samples was tested by using samples from periodontal tooth pockets and from the chemocline of a hypersaline cyanobacterial mat from Solar Lake (Sinai, Egypt). Consistent with previous studies, SRP-PhyloChip indicated the occurrence of Desulfomicrobium spp. in the tooth pockets and the presence of Desulfonema- and Desulfomonile-like SRPs (together with other SRPs) in the chemocline of the mat. The SRP-PhyloChip results were confirmed by several DNA microarray-independent techniques, including specific PCR amplification, cloning, and sequencing of SRP 16S rRNA genes and the genes encoding the dissimilatory (bi)sulfite reductase (dsrAB).
Bacterial community composition and function in sewage treatment systems
2002 - Curr. Opin. Biotechnol., 13: 218-227
Abstract:
The application of modern molecular techniques has led to the identification, in situ quantification, and partial ecophysiological characterisation of bacteria responsible for bulking and foaming or for nutrient removal in sewage treatment systems. Unexpectedly, previously unrecognised, yet uncultured bacteria were demonstrated to catalyse nitrogen and phosphorous removal in activated-sludge and biofilm reactors. These findings provide the basis for the development of novel concepts for improving the efficiency and functional stability of waste water treatment systems.
The microbial community composition of a nitrifying-denitrifying activated sludge from an industrial sewage treatment plant analyzed by the full-cycle rRNA approach
2002 - Syst. Appl. Microbiol., 25: 84-99
Abstract:
The composition of the microbial community present in the nitrifying-denitrifying activated sludge of an industrial wastewater treatment plant connected to a rendering facility was investigated by the full-cycle rRNA approach. After DNA extraction using three different methods, 94 almost full-length 16S rRNA gene clones were retrieved and analyzed phylogenetically. 59% of the clones were affiliated with the Proteobacteria and clustered with the beta- (29 clones), alpha- (24), and delta-class (2 clones), respectively. 15 clones grouped within the green nonsulfur (GNS) bacteria and 11 clones belonged to the Planctomycetes. The Verrucomicrobia, Acidobacteria, Nitrospira, Bacteroidetes, Firmicutes and Actinobacteria were each represented by one to five clones. Interestingly, the highest 'species richness' [measured as number of operational taxonomic units (OTUs)] was found within the alpha-class of Proteobacteria, followed by the Planctomycetes, the beta-class of Proteobacteria, and the GNS-bacteria. The microbial community composition of the activated sludge was determined quantitatively by using 36 group-, subgroup-, and OTU-specific rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization (FISH), confocal laser scanning microscopy and digital image analysis. 89% of all bacteria detectable by FISH with a bacterial probe set could be assigned to specific divisions. Consistent with the 16S rRNA gene library data, members of the beta-class of Proteobacteria dominated the microbial community and represented almost half of the biovolume of all bacteria detectable by FISH. Within the beta-class, 98% of the cells could be identified by the application of genus- or OTU-specific probes demonstrating a high in situ abundance of bacteria related to Zoogloea and Azoarcus sensu lato. Taken together, this study provides the first encompassing, high-resolution insight into the in situ composition of the microbial community present in a full-scale, industrial wastewater treatment plant.
Book chapters and other publications
Phylogenetic microarrays for cultivation-independent identification and metabolic characterization of microorganisms in complex samples
2011 - 187-206. in PCR mutation detection protocols. Methods in Molecular Biolology (Vol. 688). (Theophilus BDM, Rapley R). Springer, Dordrecht Heidelberg London New York
Abstract:
High-throughput sequencing and hybridization technologies promise new insights into the natural diversity and dynamics of microorganisms. Among these new technologies are phylogenetic oligonucleotide microarrays (phylochips) that depend on the standard molecules for taxonomic and environmental studies of microorganisms: the ribosomal RNAs and their encoding genes. The beauty of phylochip hybridization is that a sample can be analyzed with hundreds to thousands of rRNA (gene)-targeted probes simultaneously, lending itself to the efficient diagnosis of many target organisms in many samples. An emerging application of phylochips is the highly parallel analysis of structure-function relationships of microbial community members by employing in vivo substrate-mediated isotope labeling of rRNA (via the isotope array approach). This chapter provides an introduction to phylochip and isotope array analysis and detailed wet-lab protocols for preparation, labeling, and hybridization of target nucleic acids.
Probing identity and physiology of uncultured microorganisms with isotope labeling techniques
2010 - 127-146. in Geomicrobiology: Molecular and Environmental Perspective. (Loy A, Barton LL, Mandl M). Springer, Dordrecht, Heidelberg, London, New York
Microarrays for studying the composition and function of microbial communities
2010 - 397-411. in The microbiology of activated sludge. (Nielsen PH, Seviour RJ). IWA Publishing, London, UK
Preface
2010 - in Geomicrobiology: Molecular and Environmental Perspective. (Barton LL, Mandl M, Loy A). Springer, Dordrecht Heidelberg London, New York
Abstract:
This book is an interdisciplinary review of recent developments in topics including origin of life, microbial-mineral interactions, and microbial processes functioning in marine and terrestrial environments. A major component of this book addresses molecular techniques to evaluate microbial evolution and assess relationships of microbes in complex, natural communities. The function of microbial community members and their possible geological impact are evaluated from a perspective of (meta)genomics, (meta)proteomics, and isotope labeling. As well as summarizing current knowledge in various areas, it also reveals unresolved questions that require future investigations. These chapters enhance our fundamental knowledge of geomicrobiology that contributes to the exploitation of microbial functions in mineral and environmental biotechnology applications. Authors have provided skillful reviews and outlined unique perspectives on environmental microorganisms and their related processes.
Evolution and ecology of microbes dissimilating sulfur compounds: Insights from siroheme sulfite reductases
2008 - 46-59. in Microbial Sulfur Metabolism. (Friedrich C, Dahl C). Springer, New York
Abstract:
Sulfur microorganisms have been thriving on Earth since the dawn of life and are still of central importance for the functioning of modern ecosystems. Here, we summarize the current perception of the evolution of dissimilatory siroheme sulfite reductases (DSRs), antique key enzymes in the energy metabolism of sulfur microbes. We further give recent examples of the diversity and ecology of uncultured sulfur-dissimilating microorganisms; unprecedented insights that were only made possible by exploiting DSR-encoding genes as molecular markers in environmental surveys.
Molecular strategies for studies of natural populations of sulphate-reducing microorganisms
2007 - 39-117. in Sulfate-reducing bacteria: Environmental and engineered systems. (Barton LL, Hamilton WA). Cambridge University Press, Cambridge, UK
Applications of nucleic acid microarrays in soil microbial ecology
2006 - 18-41. in Molecular approaches to soil, rhizosphere and plant microorganism analysis. (Cooper JE, Rao JR). CABI Publishing, Wallingford, Oxfordshire, UK
Abstract:
This chapter introduces the general methodological principles of DNA microarrays and highlight crucial steps in developing and applying this hybridization format for the analysis of complex microbial communities in the environment. Special emphasis is given to soil habitats. Microarrays for microbial community analysis have been classified into three main categories depending on the combination of probe types and target molecules exploited: (i) community genome arrays; (ii) rRNA-based oligonucleotide microarrays (PhyloChips, phylogenetic oligonucleotide arrays); and (iii) functional gene arrays. Benefits and caveats of these three microarrays are illustrated and discussed. The application of microarray technology is not restricted to identification of genes/microorganisms in the environment, but additionally has great potential for ecophysiological characterization of microbial populations.
Oligonucleotide microarrays in microbial diagnostics
2005 - 1-8. in Encyclopedia of medical genomics and proteomics. (Fuchs J, Podda M). Marcel Dekker Inc., New York, USAAnalyse komplexer mikrobieller Lebensgemeinschaften mit phylogenetischen Mikroarrays
2004 - BioSpektrum, 6: 745-747
Activated Sludge - Molecular techniques for determining community composition
2002 - 26-43. in The Encyclopedia of Environmental Microbiology. (Bitton G). Wiley - New York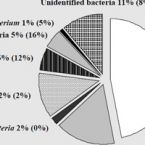

Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at