Publications
Publications in peer reviewed journals
The Fish Pathogen "Candidatus Clavichlamydia salmonicola"-A Missing Link in the Evolution of Chlamydial Pathogens of Humans.
2023 - Genome Biol Evol, 8: evad147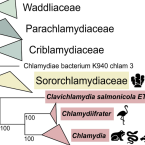
Abstract:
Chlamydiae like Chlamydia trachomatis and Chlamydia psittaci are well-known human and animal pathogens. Yet, the chlamydiae are a much larger group of evolutionary ancient obligate intracellular bacteria that includes predominantly symbionts of protists and diverse animals. This makes them ideal model organisms to study evolutionary transitions from symbionts in microbial eukaryotes to pathogens of humans. To this end, comparative genome analysis has served as an important tool. Genome sequence data for many chlamydial lineages are, however, still lacking, hampering our understanding of their evolutionary history. Here, we determined the first high-quality draft genome sequence of the fish pathogen "Candidatus Clavichlamydia salmonicola", representing a separate genus within the human and animal pathogenic Chlamydiaceae. The "Ca. Clavichlamydia salmonicola" genome harbors genes that so far have been exclusively found in Chlamydia species suggesting that basic mechanisms important for the interaction with chordate hosts have evolved stepwise in the history of chlamydiae. Thus, the genome sequence of "Ca. Clavichlamydia salmonicola" allows to constrain candidate genes to further understand the evolution of chlamydial virulence mechanisms required to infect mammals.
Colocalization and potential interactions of and chlamydiae in microbial aggregates of the coral Pocillopora acuta
2023 - Sci Adv, 20: eadg0773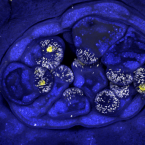
Abstract:
Corals are associated with a variety of bacteria, which occur in the surface mucus layer, gastrovascular cavity, skeleton, and tissues. Some tissue-associated bacteria form clusters, termed cell-associated microbial aggregates (CAMAs), which are poorly studied. Here, we provide a comprehensive characterization of CAMAs in the coral . Combining imaging techniques, laser capture microdissection, and amplicon and metagenome sequencing, we show that (i) CAMAs are located in the tentacle tips and may be intracellular; (ii) CAMAs contain (Gammaproteobacteria) and (Chlamydiota) bacteria; (iii) may provide vitamins to its host and use secretion systems and/or pili for colonization and aggregation; (iv) and occur in distinct, but adjacent, CAMAs; and (v) may receive acetate and heme from neighboring . Our study provides detailed insight into coral endosymbionts, thereby improving our understanding of coral physiology and health and providing important knowledge for coral reef conservation in the climate change era.
Gene gain facilitated endosymbiotic evolution of Chlamydiae.
2023 - Nat Microbiol, 1: 40-54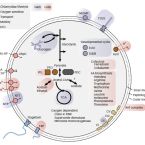
Abstract:
Chlamydiae is a bacterial phylum composed of obligate animal and protist endosymbionts. However, other members of the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum are primarily free living. How Chlamydiae transitioned to an endosymbiotic lifestyle is still largely unresolved. Here we reconstructed Planctomycetes-Verrucomicrobia-Chlamydiae species relationships and modelled superphylum genome evolution. Gene content reconstruction from 11,996 gene families suggests a motile and facultatively anaerobic last common Chlamydiae ancestor that had already gained characteristic endosymbiont genes. Counter to expectations for genome streamlining in strict endosymbionts, we detected substantial gene gain within Chlamydiae. We found that divergence in energy metabolism and aerobiosis observed in extant lineages emerged later during chlamydial evolution. In particular, metabolic and aerobic genes characteristic of the more metabolically versatile protist-infecting chlamydiae were gained, such as respiratory chain complexes. Our results show that metabolic complexity can increase during endosymbiont evolution, adding an additional perspective for understanding symbiont evolutionary trajectories across the tree of life.
Ecology and evolution of chlamydial symbionts of arthropods
2022 - ISME Commun., 2: 45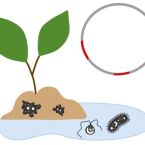
Abstract:
The phylum Chlamydiae consists of obligate intracellular bacteria including major human pathogens and diverse environmental representatives. Here we investigated the Rhabdochlamydiaceae, which is predicted to be the largest and most diverse chlamydial family, with the few described members known to infect arthropod hosts. Using published 16S rRNA gene sequence data we identified at least 388 genus-level lineages containing about 14 051 putative species within this family. We show that rhabdochlamydiae are mainly found in freshwater and soil environments, suggesting the existence of diverse, yet unknown hosts. Next, we used a comprehensive genome dataset including metagenome assembled genomes classified as members of the family Rhabdochlamydiaceae, and we added novel complete genome sequences of Rhabdochlamydia porcellionis infecting the woodlouse Porcellio scaber, and of 'Candidatus R. oedothoracis' associated with the linyphiid dwarf spider Oedothorax gibbosus. Comparative analysis of basic genome features and gene content with reference genomes of well-studied chlamydial families with known host ranges, namely Parachlamydiaceae (protist hosts) and Chlamydiaceae (human and other vertebrate hosts) suggested distinct niches for members of the Rhabdochlamydiaceae. We propose that members of the family represent intermediate stages of adaptation of chlamydiae from protists to vertebrate hosts. Within the genus Rhabdochlamydia, pronounced genome size reduction could be observed (1.49-1.93 Mb). The abundance and genomic distribution of transposases suggests transposable element expansion and subsequent gene inactivation as a mechanism of genome streamlining during adaptation to new hosts. This type of genome reduction has never been described before for any member of the phylum Chlamydiae. This study provides new insights into the molecular ecology, genomic diversity, and evolution of representatives of one of the most divergent chlamydial families.
Comparison of genovars and Chlamydia trachomatis infection loads in ocular samples from children in two distinct cohorts in Sudan and Morocco.
2021 - PLoS Negl Trop Dis, 8: e0009655
Abstract:
Trachoma is a blinding disease caused by repeated conjunctival infection with different Chlamydia trachomatis (Ct) genovars. Ct B genovars have been associated with more severe trachoma symptoms. Here, we investigated associations between Ct genovars and bacterial loads in ocular samples from two distinct geographical locations in Africa, which are currently unclear. We tested ocular swabs from 77 Moroccan children (28 with trachomatous inflammation-follicular (TF) and 49 healthy controls), and 96 Sudanese children (54 with TF and 42 healthy controls) with a Ct-specific real-time polymerase chain reaction (PCR) assay. To estimate bacterial loads, Ct-positive samples were further processed by multiplex real-time qPCR to amplify the chromosomal outer membrane complex B and plasmid open reading frame 2 of Ct. Genotyping was performed by PCR-based amplification of the outer membrane protein A gene (~1120 base pairs) of Ct and Sanger sequencing. Ct-positivities among the Moroccan and Sudanese patient groups were 60·7% and 31·5%, respectively. Significantly more Sudanese patients than Moroccan patients were genovar A-positive. In contrast, B genovars were significantly more prevalent in Moroccan patients than in Sudanese patients. Significantly higher Ct loads were found in samples positive for B genovars (598·596) than A genovar (51·005). Geographical differences contributed to the distributions of different ocular Ct genovars. B genovars may induce a higher bacterial load than A genovars in trachoma patients. Our findings emphasize the importance of conducting broader studies to elucidate if the noted difference in multiplication abilities are genovar and/or endemicity level dependent.
Pangenomics reveals alternative environmental lifestyles among chlamydiae
2021 - Nature Commun, 12: 4021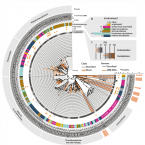
Abstract:
Chlamydiae are highly successful strictly intracellular bacteria associated with diverse eukaryotic hosts. Here we analysed metagenome-assembled genomes of the “Genomes from Earth’s Microbiomes” initiative from diverse environmental samples, which almost double the known phylogenetic diversity of the phylum and facilitate a highly resolved view at the chlamydial pangenome. Chlamydiae are defined by a relatively large core genome indicative of an intracellular lifestyle, and a highly dynamic accessory genome of environmental lineages. We observe chlamydial lineages that encode enzymes of the reductive tricarboxylic acid cycle and for light-driven ATP synthesis. We show a widespread potential for anaerobic energy generation through pyruvate fermentation or the arginine deiminase pathway, and we add lineages capable of molecular hydrogen production. Genome-informed analysis of environmental distribution revealed lineage-specific niches and a high abundance of chlamydiae in some habitats. Together, our data provide an extended perspective of the variability of chlamydial biology and the ecology of this phylum of intracellular microbes.
Coevolving plasmids drive gene flow and genome plasticity in host-associated intracellular bacteria
2021 - Curr Biol, 2: 346-357.e3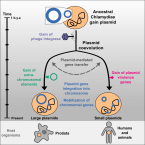
Abstract:
Plasmids are important in microbial evolution and adaptation to new environments. Yet, carrying a plasmid can be costly, and long-term association of plasmids with their hosts is poorly understood. Here, we provide evidence that the Chlamydiae, a phylum of strictly host-associated intracellular bacteria, have coevolved with their plasmids since their last common ancestor. Current chlamydial plasmids are amalgamations of at least one ancestral plasmid and a bacteriophage. We show that the majority of plasmid genes are also found on chromosomes of extant chlamydiae. The most conserved plasmid gene families are predominantly vertically inherited, while accessory plasmid gene families show significantly increased mobility. We reconstructed the evolutionary history of plasmid gene content of an entire bacterial phylum over a period of around one billion years. Frequent horizontal gene transfer and chromosomal integration events illustrate the pronounced impact of coevolution with these extrachromosomal elements on bacterial genome dynamics in host-dependent microbes.
Chlamydiae in the Environment.
2020 - Trends Microbiol, 11: 877-888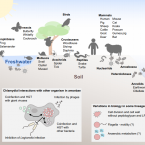
Abstract:
Chlamydiae have been known for more than a century as major pathogens of humans. Yet they are also found ubiquitously in the environment where they thrive within protists and in an unmatched wide range of animals. This review summarizes recent advances in understanding chlamydial diversity and distribution in nature. Studying these environmental chlamydiae provides a novel perspective on basic chlamydial biology and evolution. A picture is beginning to emerge with chlamydiae representing one of the evolutionarily most ancient and successful groups of obligate intracellular bacteria.
Detection of Chlamydiaceae and Chlamydia-like organisms on the ocular surface of children and adults from a trachoma-endemic region
2018 - Sci Rep, 1: 7432
Abstract:
Trachoma, the leading infectious cause of blindness, is caused by Chlamydia trachomatis (Ct), a bacterium of the phylum Chlamydiae. Recent investigations revealed the existence of additional families within the phylum Chlamydiae, also termed Chlamydia-like organisms (CLOs). In this study, the frequency of Ct and CLOs was examined in the eyes of healthy Sudanese (control) participants and those with trachoma (case). We tested 96 children (54 cases and 42 controls) and 93 adults (51 cases and 42 controls) using broad-range Chlamydiae and Ct-specific (omcB) real-time PCR. Samples positive by broad-range Chlamydiae testing were subjected to DNA sequencing. Overall Chlamydiae prevalence was 36%. Sequences corresponded to unclassified and classified Chlamydiae. Ct infection rate was significantly higher in children (31.5%) compared to adults (0%) with trachoma (p < 0.0001). In general, 21.5% of adults and 4.2% of children tested positive for CLOs (p = 0.0003). Our findings are consistent with previous investigations describing the central role of Ct in trachoma among children. This is the first study examining human eyes for the presence of CLOs. We found an age-dependent distribution of CLO DNA in human eyes with significantly higher positivity in adults. Further studies are needed to understand the impact of CLOs in trachoma pathogenicity and/or protection.
Unexpected genomic features in widespread intracellular bacteria: evidence for motility of marine chlamydiae.
2017 - ISME J, 10: 2334-2344
Abstract:
Chlamydiae are obligate intracellular bacteria comprising important human pathogens and symbionts of protists. Molecular evidence indicates a tremendous diversity of chlamydiae particularly in marine environments, yet our current knowledge is based mainly on terrestrial representatives. Here we provide first insights into the biology of marine chlamydiae representing three divergent clades. Our analysis of single-cell amplified genomes revealed hallmarks of the chlamydial lifestyle, supporting the ancient origin of their characteristic developmental cycle and major virulence mechanisms. Surprisingly, these chlamydial genomes encode a complete flagellar apparatus, a previously unreported feature. We show that flagella are an ancient trait that was subject to differential gene loss among extant chlamydiae. Together with a chemotaxis system, these marine chlamydiae are likely motile, with flagella potentially playing a role during host cell infection. This study broadens our view on chlamydial biology and indicates a largely underestimated potential to adapt to different hosts and environments.
Draft genome sequence of 'Candidatus Hepatoplasma crinochetorum Ps' – a bacterial symbiont in the hepatopancreas of the terrestrial isopod Porcellio scaber
2015 - Genome Announc, 3: e00674-15
Abstract:
'Candidatus Hepatoplasma crinochetorum Ps' is an extracellular symbiont residing in the hepatopancreas of the terrestrial isopod Porcellio scaber. Its genome is highly similar to that of the close relative 'Cand. Hepatoplasma crinochetorum Av' from Armadillidium vulgare. However, instead of a CRISPR/Cas system it encodes a type I restriction modification system.
Tracing the primordial Chlamydiae: extinct parasites of plants?
2014 - Trends Plant Sci., 19: 36-43
Abstract:
Chlamydiae are obligate intracellular bacteria found as symbionts and pathogens in a wide range of eukaryotes, including protists, invertebrates, and vertebrates. It was recently proposed that an ancient chlamydial symbiont facilitated the establishment of primary plastids in a tripartite symbiosis with cyanobacteria and early eukaryotes. In this review, we summarize recent advances in understanding of the lifestyle and the evolutionary history of extant Chlamydiae. We reconstruct and describe key features of the ancient chlamydial symbiont. We propose that it was already adapted to an intracellular lifestyle before the emergence of Archaeplastida, and that several observations are compatible with an essential contribution of Chlamydiae to the evolution of algae and plants.
Massive expansion of ubiquitination-related gene families within the Chlamydiae
2014 - Mol Biol Evol., 31: 2890-904Abstract:
Gene loss, gain, and transfer play an important role in shaping the genomes of all organisms; however, the interplay of these processes in isolated populations, such as in obligate intracellular bacteria, is less understood. Despite a general trend towards genome reduction in these microbes, our phylogenomic analysis of the phylum Chlamydiae revealed that within the family Parachlamydiaceae, gene family expansions have had pronounced effects on gene content. We discovered that the largest gene families within the phylum are the result of rapid gene birth-and-death evolution. These large gene families are comprised of members harboring eukaryotic-like ubiquitination-related domains, such as F-box and BTB-box domains, marking the largest reservoir of these proteins found among bacteria. A heterologous type III secretion system assay suggests that these proteins function as effectors manipulating the host cell. The large disparity in copy number of members in these families between closely related organisms suggests that nonadaptive processes might contribute to the evolution of these gene families. Gene birth-and-death evolution in concert with genomic drift might represent a previously undescribed mechanism by which isolated bacterial populations diversify.
Bacteriocyte-associated gammaproteobacterial symbionts of the Adelges nordmannianae/piceae complex (Hemiptera: Adelgidae)
2012 - ISME J., 6: 384-396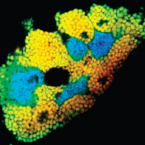
Abstract:
Adelgids (Insecta: Hemiptera: Adelgidae) are known as severe pests of various conifers in North America, Canada, Europe and Asia. Here we present the first molecular identification of bacteriocyte-associated symbionts in these plant sap-sucking insects. Three geographically distant populations of members of the Adelges nordmannianae/piceae complex, identified based on coI and ef1alpha gene sequences, were investigated. Electron and light microscopy revealed two morphologically different endosymbionts, coccoid or polymorphic, that are located in distinct bacteriocytes. Phylogenetic analyses of their 16S and 23S rRNA gene sequences assigned both symbionts to novel lineages within the Gammaproteobacteria sharing less than 92% 16S rRNA sequence similarity with each other and showing no close relationship with known symbionts of insects. Their identity and intracellular location were confirmed by fluorescence in situ hybridisation, and the names ‘Candidatus Steffania adelgidicola’ and ‘Candidatus Ecksteinia adelgidicola’ are proposed for tentative classification. Both symbionts were present in all individuals of all investigated populations and in different adelgid life stages including eggs, suggesting vertical transmission from mother to offspring. An 85 kb genome fragment of ‘Candidatus Steffania adelgidicola’ was reconstructed based on a metagenomic library created from purified symbionts. Genomic features including the frequency of pseudogenes, the average length of intergenic regions and the presence of several genes, which are absent in other long-term obligate symbionts, suggested that ‘Candidatus Steffania adelgidicola’ is an evolutionary young bacteriocyte-associated symbiont, which has been acquired after the diversification of adelgids from their aphid sister group.
Unity in variety - the pan-genome of the Chlamydiae
2011 - Mol. Biol. Evol., 28: 3253-3270Abstract:
Chlamydiae are evolutionarily well-separated bacteria that live exclusively within eukaryotic host cells. They include important human pathogens such as Chlamydia trachomatis as well as symbionts of protozoa. As these bacteria are experimentally challenging and genetically intractable, our knowledge about them is still limited. In this study, we obtained the genome sequences of Simkania negevensis Z, Waddlia chondrophila 2032/99 and Parachlamydia acanthamoebae UV-7. This enabled us to perform the first comprehensive comparative and phylogenomic analysis of representative members of four major families of the Chlamydiae, including the Chlamydiaceae. We identified a surprisingly large core gene set present in all genomes and a high number of diverse accessory genes in those Chlamydiae that do not primarily infect humans or animals, including a chemosensory system in P. acanthamoebae and a type IV secretion system. In S. negevensis, the type IV secretion system is encoded on a large conjugative plasmid (pSn, 132 kb). Phylogenetic analyses suggested that a plasmid similar to the S. negevensis plasmid was originally acquired by the last common ancestor of all four families and that it was subsequently reduced, integrated into the chromosome, or lost during diversification, ultimately giving rise to the extant virulence-associated plasmid of pathogenic chlamydiae. Other virulence factors, including a type III secretion system, are conserved among the Chlamydiae to variable degrees, and together with differences in the composition of the cell wall, reflect adaptation to different host cells including convergent evolution among the four chlamydial families. Phylogenomic analysis focusing on chlamydial proteins with homology to plant proteins provided evidence for the acquisition of 53 chlamydial genes by a plant progenitor, lending further support for the hypothesis of an early interaction between a chlamydial ancestor and the primary photosynthetic eukaryote.
Chlamydia-like bacteria in respiratory samples of community-acquired pneumonia patients
2008 - FEMS Microbiol. Lett., 281: 198-202
Abstract:
Chlamydia-like bacteria, obligate intracellular relatives of Chlamydia trachomatis and Chlamydophila pneumoniae, are widely distributed in nature. Using a two-step nested and semi-nested PCR approach targeting the 16S rRNA gene, we found DNA of chlamydia-like bacteria in respiratory samples from patients with community-acquired pneumonia. Four out of 387 cases (1.03%) tested positive if only sequences showing less than 99.9% 16S rRNA sequence similarity to known chlamydiae were considered. These included for the first time Protochlamydia amoebophila, Waddlia chondrophila, and ‘Candidatus Rhabdochlamydia porcellionis’-related sequences. This study extends previous findings suggesting an association of chlamydia-like bacteria with respiratory disease, but a causal link between these microorganisms and respiratory tract infections has yet to be established.
Deciphering the evolution and metabolism of an anammox bacterium from a community genome
2006 - Nature, 440: 790-794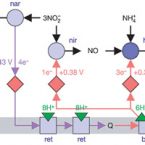
Abstract:
Anaerobic ammonium oxidation (anammox) has become a main focus in oceanography and wastewater treatment. It is also the nitrogen cycle's major remaining biochemical enigma. Among its features, the occurrence of hydrazine as a free intermediate of catabolism, the biosynthesis of ladderane lipids and the role of cytoplasm differentiation are unique in biology. Here we use environmental genomics the reconstruction of genomic data directly from the environment to assemble the genome of the uncultured anammox bacterium Kuenenia stuttgartiensis from a complex bioreactor community. The genome data illuminate the evolutionary history of the Planctomycetes and allow us to expose the genetic blueprint of the organism's special properties. Most significantly, we identified candidate genes responsible for ladderane biosynthesis and biological hydrazine metabolism, and discovered unexpected metabolic versatility.
'Candidatus Protochlamydia amoebophila', an endosymbiont of Acanthamoeba spp
2005 - Int. J. Syst. Evol. Microbiol., 55: 1863-1866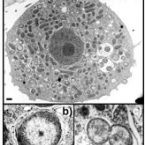
Abstract:
The obligately intracellular coccoid bacterium UWE25, a symbiont of Acanthamoeba spp., was previously identified as being related to chlamydiae based upon the presence of a chlamydia-like developmental cycle and its 16S rRNA gene sequence. Analysis of its complete genome sequence demonstrated that UWE25 shows many characteristic features of chlamydiae, including dependency on host-derived metabolites, composition of the cell envelope and the ability to thrive as an energy parasite within the cells of its eukaryotic host. Phylogenetic analysis of 44 ribosomal proteins further confirmed the affiliation of UWE25 to the 'Chlamydiae'. Within this phylum, UWE25 could be assigned to the family Parachlamydiaceae based on comparative analyses of the 16S rRNA, 23S rRNA and endoribonuclease P RNA genes. The distinct dissimilarities from its closest relative, Parachlamydia acanthamoebae Bn(9)(T) (7.1, 9.7 and 28.8%, respectively), observed in this analysis justify its classification in a new genus. Therefore, the name 'Candidatus Protochlamydia amoebophila' is proposed for the designation of the Acanthamoeba sp. symbiont UWE25 (=ATCC PRA-7).
Recovery of an environmental chlamydia strain from activated sludge by co-cultivation with Acanthamoeba sp
2005 - Microbiology, 151: 301-309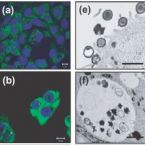
Abstract:
Chlamydiae are a unique group of obligate intracellular bacteria comprising important pathogens of vertebrates as well as symbionts of free-living amoebae. Although there is ample molecular evidence for a huge diversity and wide distribution of chlamydiae in nature, environmental chlamydiae are currently represented by only few isolates. This paper reports the recovery of a novel environmental chlamydia strain from activated sludge by co-cultivation with Acanthamoeba sp. The recovered environmental chlamydia strain UV-7 showed the characteristic morphology of chlamydial developmental stages as revealed by electron microscopy and was identified as a new member of the family Parachlamydiaceae (98.7 % 16S rRNA sequence similarity to Parachlamydia acanthamoebae). Infection studies suggested that Parachlamydia sp. UV-7 is not confined to amoeba hosts but is also able to invade mammalian cells. These findings outline a new straightforward approach to retrieving environmental chlamydiae from nature without prior, tedious isolation and cultivation of their natural host cells, and lend further support to suggested implications of environmental chlamydiae for public health.
Chlamydial endocytobionts of free-living amoebae differentially affect the growth rate of their hosts
2004 - Europ. J. Protist., 40: 57-60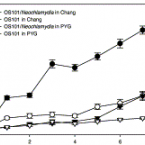
A candidate NAD+ transporter in an intracellular bacterial symbiont related to chlamydiae
2004 - Nature, 432: 622-625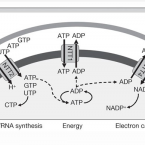
Abstract:
Bacteria living within eukaryotic cells can be essential for the survival or reproduction of the host but in other cases are among the most successful pathogens. Environmental Chlamydiae, including strain UWE25, thrive as obligate intracellular symbionts within protozoa; are recently discovered relatives of major bacterial pathogens of humans; and also infect human cells. Genome analysis of UWE25 predicted that this symbiont is unable to synthesize the universal electron carrier nicotinamide adenine dinucleotide (NAD+). Compensation of limited biosynthetic capacity in intracellular bacteria is usually achieved by import of primary metabolites. Here, we report the identification of a candidate transporter protein from UWE25 that is highly specific for import of NAD+ when synthesized heterologously in Escherichia coli. The discovery of this candidate NAD+/ADP exchanger demonstrates that intact NAD+ molecules can be transported through cytoplasmic membranes. This protein acts together with a newly discovered nucleotide transporter and an ATP/ADP translocase, and allows UWE25 to exploit its host cell by means of a sophisticated metabolic parasitism.
Illuminating the evolutionary history of chlamydiae
2004 - Science, 304: 728-730- Supplementary material [PDF]
- Press release [PDF]
- Press release (German) [PDF]
- Movie showing environmental chlamydia UWE25 within its amoeba host cells. AAAS/Science. [Windows Media Player]
- Movie showing environmental chlamydia UWE25 within its amoeba host cells. AAAS/Science.[Quicktime]
- Environmental chlamydia genome database
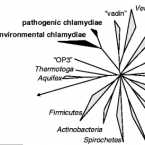
Abstract:
Chlamydiae are the major cause of preventable blindness and sexually transmitted disease. Genome analysis of a chlamydia-related symbiont of free-living amoebae revealed that it is twice as large as any of the pathogenic chlamydiae and had few signs of recent lateral gene acquisition. We showed that about 700 million years ago the last common ancestor of pathogenic and symbiotic chlamydiae was already adapted to intracellular survival in early eukaryotes and contained many virulence factors found in modern pathogenic chlamydiae, including a type III secretion system. Ancient chlamydiae appear to be the originators of mechanisms for the exploitation of eukaryotic cells.
ATP/ADP translocases: A common feature of obligate intracellular amoebal symbionts related to chlamydiae and rickettsiae
2004 - J. Bacteriol., 186: 683-691
Abstract:
ATP/ADP translocases catalyze the highly specific transport of ATP across a membrane in an exchange mode with ADP. Such unique transport proteins are employed by plant plastids and have among the prokaryotes so far only been identified in few obligate intracellular bacteria belonging to the Chlamydiales and the Rickettsiales. In this study, 12 phylogenetically diverse bacterial endosymbionts of free-living amoebae and paramecia were screened for the presence of genes encoding ATP/ADP transport proteins. The occurrence of ATP/ADP translocase genes was found to be restricted to endosymbionts related to rickettsiae and chlamydiae. We showed that the ATP/ADP transport protein of the Parachlamydia-related endosymbiont of Acanthamoeba sp. strain UWE25, a recently identified relative of the important human pathogens Chlamydia trachomatis and Chlamydophila pneumoniae, is functional when expressed in the heterologous host Escherichia coli and demonstrated the presence of transcripts during the chlamydial developmental cycle. These findings indicate that the interaction between Parachlamydia-related endosymbionts and their amoeba hosts concerns energy parasitism similar to the interaction between pathogenic chlamydiae and their human host cells. Phylogenetic analysis of all known ATP/ADP translocases indicated that the genes encoding ATP/ADP translocases originated from a chlamydial ancestor and were, after an ancient gene duplication, transferred horizontally to rickettsiae and plants.
Book chapters and other publications
Draft genome sequences of Chlamydiales bacterium STE3 and Neochlamydia sp. AcF84, endosymbionts of Acanthamoeba spp.
2020 - Microbiol Resour Announc, 9: e00220-20
Abstract:
Chlamydiales bacterium STE3 and Neochlamydia sp. strain AcF84 are obligate intracellular symbionts of Acanthamoeba spp. isolated from the biofilm of a littoral cave wall and gills from striped tiger leaf fish, respectively. We report the draft genome sequences of these two environmental chlamydiae affiliated with the family Parachlamydiaceae.
Environmental chlamydia genomics
2006 - 25-44. in Chlamydia: genomics and pathogenesis. (Bavoil PM, Wyrick PB). Horizon Scientific Press, Norfolk
Abstract:
The discovery of chlamydia-related bacteria in the environment dramatically changed our perception of chlamydial diversity and their environmental occurrence. These so-called environmental chlamydiae display the unique chlamydial developmental cycle and mainly live as symbionts of free-living amoebae. Although there are some indications that environmental chlamydiae might also be able to infect humans, their pathogenic potential is still unclear. The first complete genome analysis of an environmental chlamydia strain, Protochlamydia amoebophila UWE25, provided novel insights into its biology, which, because of its larger genome compared to the Chlamydiaceae, opened a window on the genetic make-up of the chlamydial ancestor and the evolution of chlamydial virulence. The last common ancestor of the environmental chlamydiae and Chlamydiaceae was already adapted to intracellular survival in early eukaryotes, but was less dependent on its host cell. Several virulence factors required by Chlamydiaceae for interaction with their hosts evolved from genes of the last common ancestor of the environmental chlamydiae and the Chlamydiaceae.
Chlamydien als Symbionten frei lebender Amöben
2005 - BioSpektrum, 4: 393-394Die Evolution der Chlamydien - Einblicke in die Entwicklungsgeschichte bedeutender bakterieller Krankheitserreger aus einer genomischen Perspektive
2004 - GenomXPress, 2: 13-14
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at