Publications
Publications in peer reviewed journals
The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
2024 - Acta Obstet Gynecol Scand, in press
Abstract:
Changes within the maternal microbiome during the last trimester of pregnancy and the determinants of the subsequent neonatal microbiome establishment after delivery by elective cesarean section are described.
Maternal vaginal and rectal microbiome samples were collected in the last trimester and before cesarean section; intrauterine cavity, placenta, neonatal buccal mucosa, skin, and meconium samples were obtained at birth; neonatal sample collection was repeated 2-3 days postnatally. Microbial community composition was analyzed by 16S rRNA gene amplicon sequencing. Relative abundance measurements of amplicon sequencing variants and sum counts at higher taxonomic levels were compared to test for significant overlap or differences in microbial community compositions.
gov ID: NCT04489056.
A total of 30 mothers and their neonates were included with available microbiome samples for all maternal, intrauterine cavity and placenta samples, as well as for 18 of 30 neonates. The composition of maternal vaginal and rectal microbiomes during the last trimester of healthy pregnancies did not significantly change (permutational multivariate analysis of variance [PERMANOVA], p > 0.05). No robust microbial signature was detected in the intrauterine cavity, placenta, neonatal buccal mucosa, skin swabs, or meconium samples collected at birth. After birth, the neonatal microbiome was rapidly established, and significantly different microbial communities were detectable 2-3 days postnatally in neonate buccal mucosa and stool samples (PERMANOVA, p < 0.01).
Maternal vaginal and rectal microbiomes in healthy pregnancies remain stable during the third trimester. No microbial colonization of the neonate was observed before birth in healthy pregnancies. Neonatal microbiomes in infants delivered by cesarean section displayed a taxonomic composition distinct from maternal vaginal and rectal microbiomes at birth, indicating that postnatal exposure to the extrauterine environment is the driving source of initial neonatal microbiome development in this cohort.Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
2024 - Environ Sci Technol, 5: 2236-2246
Abstract:
Mycotoxins are toxic chemicals that adversely affect human health. Here, we assessed the influence of mycotoxin exposure on the longitudinal development of early life intestinal microbiota of Nigerian neonates and infants (NIs). Human biomonitoring assays based on liquid chromatography tandem mass spectrometry were applied to quantify mycotoxins in breast milk ( = 68) consumed by the NIs, their stool ( = 82), and urine samples ( = 15), which were collected longitudinally from month 1-18 postdelivery. Microbial community composition was characterized by 16S rRNA gene amplicon sequencing of stool samples and was correlated to mycotoxin exposure patterns. Fumonisin B (FB), FB, and alternariol monomethyl ether (AME) were frequently quantified in stool samples between months 6 and 18. Aflatoxin M (AFM), AME, and citrinin were quantified in breast milk samples at low concentrations. AFM, FB, and ochratoxin A were quantified in urine samples at relatively high concentrations. and / were dominant in very early life stool samples (month 1), whereas was dominant between months 3 and 6. The total mycotoxin levels in stool were significantly associated with NIs' gut microbiome composition (PERMANOVA, < 0.05). However, no significant correlation was observed between specific microbiota and the detection of certain mycotoxins. Albeit a small cohort, this study demonstrates that mycotoxins may influence early life gut microbiome composition.
Microclimate shapes the phylosymbiosis of rodent gut microbiota in Jordan's Great Rift Valley.
2023 - Front Microbiol, 1258775
Abstract:
Host phylogeny and the environment play vital roles in shaping animal microbiomes. However, the effects of these variables on the diversity and richness of the gut microbiome in different bioclimatic zones remain underexplored. In this study, we investigated the effects of host phylogeny and bioclimatic zone on the diversity and composition of the gut microbiota of two heterospecific rodent species, the spiny mouse and the house mouse , in three bioclimatic zones of the African Great Rift Valley (GRV). We confirmed host phylogeny using the sequencing method and analyzed the influence of host phylogeny and bioclimatic zone parameters on the rodent gut microbiome using high-throughput amplicon sequencing of 16S rRNA gene fragments. Phylogenetic analysis supported the morphological identification of the rodents and revealed a marked genetic difference between the two heterospecific species. We found that bioclimatic zone had a significant effect on the gut microbiota composition while host phylogeny did not. Microbial alpha diversity of heterospecific hosts was highest in the Mediterranean forest bioclimatic zone, followed by the Irano-Turanian shrubland, and was lowest in the Sudanian savanna tropical zone. The beta diversity of the two rodent species showed significant differences across the Mediterranean, Irano-Turanian, and Sudanian regions. The phyla and were highly abundant, and and were also prominent. Amplicon sequence variants (ASVs) were identified that were unique to the Sudanian bioclimatic zone. The core microbiota families recovered in this study were consistent among heterospecific hosts. However, diversity decreased in conspecific host populations found at lower altitudes in Sudanian bioclimatic zone. The composition of the gut microbiota is linked to the adaptation of the host to its environment, and this study underscores the importance of incorporating climatic factors such as elevation and ambient temperature, in empirical microbiome research and is the first to describe the rodent gut microbiome from the GRV.
Neutral and Pectic Heteropolysaccharides Isolated from Mucilage: Composition, Molecular Dimensions and Prebiotic Potential.
2023 - Int J Mol Sci, 4: in press
Abstract:
is a semi-wild cactus cultivated for its fruit. However, the cladodes are often discarded, wasting the potentially useful mucilage in them. The mucilage is composed primarily of heteropolysaccharides, characterized by their molar mass distribution, monosaccharide composition, structural features (by vibrational spectroscopy, FT IR, and atomic force microscopy, AFM), and fermentability by known saccharolytic commensal members of the gut microbiota. After fractionation with ion exchange chromatography, four polysaccharides were found: one neutral (composed mainly of galactose, arabinose, and xylose) and three acidic, with a galacturonic acid content from 10 to 35%. Their average molar masses ranged from 1.8 × 10 to 2.8 × 10 g·mol. Distinct structural features such as galactan, arabinan, xylan, and galacturonan motifs were present in the FT IR spectra. The intra- and intermolecular interactions of the polysaccharides, and their effect on the aggregation behavior, were shown by AFM. The composition and structural features of these polysaccharides were reflected in their prebiotic potential. and were not able to utilize them, whereas members of showed utilization capacity. The obtained data suggest a high economic potential for this species, with potential uses such as animal feed in arid areas, precise prebiotic, and symbiotic formulations, or as the carbon skeleton source in a green refinery. Our methodology can be used to evaluate the saccharides as the phenotype of interest, helping to guide the breeding strategy.
Gut microbiome signatures of Yorkshire Terrier enteropathy during disease and remission.
2023 - Sci Rep, 1: 4337
Abstract:
The role of the gut microbiome in developing Inflammatory Bowel Disease (IBD) in humans and dogs has received attention in recent years. Evidence suggests that IBD is associated with alterations in gut microbial composition, but further research is needed in veterinary medicine. The impact of IBD treatment on the gut microbiome needs to be better understood, especially in a breed-specific form of IBD in Yorkshire Terriers known as Yorkshire Terrier Enteropathy (YTE). This study aimed to investigate the difference in gut microbiome composition between YTE dogs during disease and remission and healthy Yorkshire Terriers. Our results showed a significant increase in specific taxa such as Clostridium sensu stricto 1, Escherichia-Shigella, and Streptococcus, and a decrease in Bacteroides, Prevotella, Alloprevotella, and Phascolarctobacterium in YTE dogs compared to healthy controls. No significant difference was found between the microbiome of dogs in remission and those with active disease, suggesting that the gut microbiome is affected beyond clinical recovery.
Pathometagenomics reveals susceptibility to intestinal infection by Morganella to be mediated by the blood group-related B4galnt2 gene in wild mice.
2023 - Gut Microbes, 1: 2164448
Abstract:
Infectious disease is widely considered to be a major driver of evolution. A preponderance of signatures of balancing selection at blood group-related genes is thought to be driven by inherent trade-offs in susceptibility to disease. B4galnt2 is subject to long-term balancing selection in house mice, where two divergent allele classes direct alternative tissue-specific expression of a glycosyltransferase in the intestine versus blood vessels. The blood vessel allele class leads to prolonged bleeding times similar to von Willebrand disease in humans, yet has been maintained for millions of years. Based on in vivo functional studies in inbred lab strains, it is hypothesized that the cost of prolonged bleeding times may be offset by an evolutionary trade-off involving susceptibility to a yet unknown pathogen(s). To identify candidate pathogens for which resistance could be mediated by B4galnt2 genotype, we here employed a novel "pathometagenomic" approach in a wild mouse population, which combines bacterial 16S rRNA gene-based community profiling with histopathology of gut tissue. Through subsequent isolation, genome sequencing and controlled experiments in lab mice, we show that the presence of the blood vessel allele is associated with resistance to a newly identified subspecies of Morganella morganii, a clinically important opportunistic pathogen. Given the increasing importance of zoonotic events, the approach outlined here may find useful application in the detection of emerging diseases in wild animal populations.
The microbiome of kidney stones and urine of patients with nephrolithiasis.
2023 - Urolithiasis, 1: 27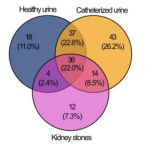
Abstract:
The incidence of nephrolithiasis is rising worldwide. Although it is a multifactorial disease, lifestyle plays a major role in its etiology. Another considerable factor could be an aberrant microbiome. In our observational single-center study, we aimed to investigate the composition of bacteria in kidney stones and urine focusing on patients with features of metabolic syndrome. Catheterized urine and kidney stones were collected prospectively from 100 consecutive patients undergoing endoscopic nephrolithotomy between 2020 and 2021 at our clinic. Microbiome composition was analyzed via 16S rRNA gene amplicon sequencing. Detection of bacteria was successful in 24% of the analyzed kidney stones. These patients had a prolonged length of stay compared to patients without verifiable bacteria in their stones (2.9 vs 1.5 days). Patients with features of metabolic syndrome were characterized by kidney stones colonized with classical gastrointestinal bacteria and displayed a significant enrichment of Enterococcaceae and Enterobacteriaceae. Stones of patients without features of metabolic syndrome characterized by Ureaplasma and Staphylococcaceae. Patients with bacteria in their kidney stones exhibit a longer length of stay, possibly due to more complex care. Patients presenting with features of metabolic syndrome displayed a distinct stone microbiome compared to metabolically fit patients. Understanding the role of bacteria in stone formation could enable targeted therapy, prevention of post-operative complications and new therapeutic strategies.
The helminth holobiont: a multidimensional host-parasite-microbiota interaction.
2022 - Trends Parasitol, in press
Abstract:
Gastrointestinal helminths have developed multiple mechanisms by which they manipulate the host microbiome to make a favorable environment for their long-term survival. While the impact of helminth infections on vertebrate host immunity and its gut microbiota is relatively well studied, little is known about the structure and functioning of microbial populations supported by metazoan parasites. Here we argue that an integrated understanding of the helminth-associated microbiome and its role in the host disease pathogenesis may facilitate the discovery of specific microbial and/or genetic patterns critical for parasite biology and subsequently pave the way for the development of alternative control strategies against parasites and parasitic disease.
Atypical enteropathogenic are associated with disease activity in ulcerative colitis.
2022 - Gut Microbes, 1: 2143218
Abstract:
With increasing urbanization and industrialization, the prevalence of inflammatory bowel diseases (IBDs) has steadily been rising over the past two decades. IBD involves flares of gastrointestinal (GI) inflammation accompanied by microbiota perturbations. However, microbial mechanisms that trigger such flares remain elusive. Here, we analyzed the association of the emerging pathogen atypical enteropathogenic (aEPEC) with IBD disease activity. The presence of diarrheagenic was assessed in stool samples from 630 IBD patients and 234 age- and sex-matched controls without GI symptoms. Microbiota was analyzed with 16S ribosomal RNA gene amplicon sequencing, and 57 clinical aEPEC isolates were subjected to whole-genome sequencing and in vitro pathogenicity experiments including biofilm formation, epithelial barrier function and the ability to induce pro-inflammatory signaling. The presence of aEPEC correlated with laboratory, clinical and endoscopic disease activity in ulcerative colitis (UC), as well as microbiota dysbiosis. In vitro, aEPEC strains induce epithelial p21-activated kinases, disrupt the epithelial barrier and display potent biofilm formation. The effector proteins and distinguish aEPEC cultured from UC and Crohn's disease patients, respectively. EspV-positive aEPEC harbor more virulence factors and have a higher pro-inflammatory potential, which is counteracted by 5-ASA. aEPEC may tip a fragile immune-microbiota homeostasis and thereby contribute to flares in UC. aEPEC isolates from UC patients display properties to disrupt the epithelial barrier and to induce pro-inflammatory signaling in vitro.
Disturbances in microbial skin recolonization and cutaneous immune response following allogeneic stem cell transfer
2022 - Leukemia, in press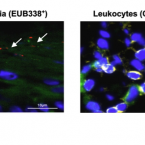
Abstract:
The composition of the gut microbiome influences the clinical course after allogeneic hematopoietic stem cell transplantation (HSCT), but little is known about the relevance of skin microorganisms. In a single-center, observational study, we recruited a cohort of 50 patients before undergoing conditioning treatment and took both stool and skin samples up to one year after HSCT. We could confirm intestinal dysbiosis following HSCT and report that the skin microbiome is likewise perturbed in HSCT-recipients. Overall bacterial colonization of the skin was decreased after conditioning. Particularly patients that developed acute skin graft-versus-host disease (aGVHD) presented with an overabundance of Staphylococcus spp. In addition, a loss in alpha diversity was indicative of aGVHD development already before disease onset and correlated with disease severity. Further, co-localization of CD45+ leukocytes and staphylococci was observed in the skin of aGVHD patients even before disease development and paralleled with upregulated genes required for antigen-presentation in mononuclear phagocytes. Overall, our data reveal disturbances of the skin microbiome as well as cutaneous immune response in HSCT recipients with changes associated with cutaneous aGVHD.
Impaired Mucosal Homeostasis in Short-Term Fiber Deprivation Is Due to Reduced Mucus Production Rather Than Overgrowth of Mucus-Degrading Bacteria.
2022 - Nutrients, 14: 3802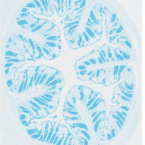
Abstract:
The gut mucosal environment is key in host health; protecting against pathogens and providing a niche for beneficial bacteria, thereby facilitating a mutualistic balance between host and microbiome. Lack of dietary fiber results in erosion of the mucosal layer, suggested to be a result of increased mucus-degrading gut bacteria. This study aimed to use quantitative analyses to investigate the diet-induced imbalance of mucosal homeostasis. Seven days of fiber-deficiency affected intestinal anatomy and physiology, seen by reduced intestinal length and loss of the colonic crypt-structure. Moreover, the mucus layer was diminished, expression decreased, and impaired mucus secretion was detected by stable isotope probing. Quantitative microbiome profiling of the gut microbiota showed a diet-induced reduction in bacterial load and decreased diversity across the intestinal tract, including taxa with fiber-degrading and butyrate-producing capabilities. Most importantly, there was little change in the absolute abundance of known mucus-degrading bacteria, although, due to the general loss of taxa, relative abundance would erroneously indicate an increase in mucus degraders. These findings underscore the importance of using quantitative methods in microbiome research, suggesting erosion of the mucus layer during fiber deprivation is due to diminished mucus production rather than overgrowth of mucus degraders.
A look beyond dietary (poly)phenols: The low molecular weight phenolic metabolites and their concentrations in human circulation.
2022 - Compr Rev Food Sci Food Saf, 5: 3931-3962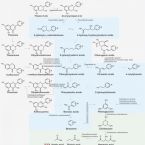
Abstract:
A large number of epidemiological studies have shown that consumption of fruits, vegetables, and beverages rich in (poly)phenols promote numerous health benefits from cardiovascular to neurological diseases. Evidence on (poly)phenols has been applied mainly to flavonoids, yet the role of phenolic acids has been largely overlooked. Such phenolics present in food combine with those resulting from gut microbiota catabolism of flavonoids and chlorogenic acids and those produced by endogenous pathways, resulting in large concentrations of low molecular weight phenolic metabolites in human circulation. Independently of the origin, in human intervention studies using diets rich in (poly)phenols, a total of 137 low molecular weight phenolic metabolites have been detected and quantified in human circulation with largely unknown biological function. In this review, we will pinpoint two main aspects of the low molecular weight phenolic metabolites: (i) the microbiota responsible for their generation, and (ii) the analysis (quali- and quantitative) in human circulation and their respective pharmacokinetics. In doing so, we aim to drive scientific advances regarding the ubiquitous roles of low molecular weight phenolic metabolites using physiologically relevant concentrations and under (patho)physiologically relevant conditions in humans.
Single-cell stable isotope probing in microbial ecology
2022 - ISME Commun, 2: 55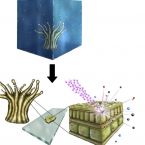
Abstract:
Environmental and host-associated microbiomes are typically diverse assemblages of organisms performing myriad activities and engaging in a network of interactions that play out in spatially structured contexts. As the sum of these activities and interactions give rise to overall microbiome function, with important consequences for environmental processes and human health, elucidating specific microbial activities within complex communities is a pressing challenge. Single-cell stable isotope probing (SC-SIP) encompasses multiple techniques that typically utilize Raman microspectroscopy or nanoscale secondary ion mass spectrometry (NanoSIMS) to enable spatially resolved tracking of isotope tracers in cells, cellular components, and metabolites. SC-SIP techniques are uniquely suited for illuminating single-cell activities in microbial communities and for testing hypotheses about cellular functions generated for example from meta-omics datasets. Here, we illustrate the insights enabled by SC-SIP techniques by reviewing selected applications in microbiology and offer a perspective on their potential for future research.
Early-life chemical exposome and gut microbiome development: African research perspectives within a global environmental health context.
2022 - Trends Microbiol, 11: 1084-1100
Abstract:
The gut microbiome of neonates, infants, and toddlers (NITs) is very dynamic, and only begins to stabilize towards the third year of life. Within this period, exposure to xenobiotics may perturb the gut environment, thereby driving or contributing to microbial dysbiosis, which may negatively impact health into adulthood. Despite exposure of NITs globally, but especially in Africa, to copious amounts and types of xenobiotics - such as mycotoxins, pesticide residues, and heavy metals - little is known about their influence on the early-life microbiome or their effects on acute or long-term health. Within the African context, the influence of fermented foods, herbal mixtures, and the delivery environment on the early-life microbiome are often neglected, despite being potentially important factors that influence the microbiome. Consequently, data on in-depth understanding of the microbiome-exposome interactions is lacking in African cohorts. Collecting and evaluating such data is important because exposome-induced gut dysbiosis could potentially favor disease progression.
Next-generation biomonitoring of the early-life chemical exposome in neonatal and infant development.
2022 - Nat Commun, 1: 2653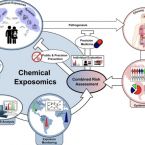
Abstract:
Exposure to synthetic and natural chemicals is a major environmental risk factor in the etiology of many chronic diseases. Investigating complex co-exposures is necessary for a holistic assessment in exposome-wide association studies. In this work, a sensitive liquid chromatography-tandem mass spectrometry approach was developed and validated. The assay enables the analysis of more than 80 highly-diverse xenobiotics in urine, serum/plasma, and breast milk; with detection limits generally in the pg-ng mL range. In plasma of extremely-premature infants, 27 xenobiotics are identified; including contamination with plasticizers, perfluorinated alkylated substances and parabens. In breast milk samples collected longitudinally over the first 211 days post-partum, 29 analytes are detected, including pyrrolizidine- and tropane alkaloids which have not been identified in this matrix before. A preliminary estimation of daily toxicant intake via breast milk is conducted. In conclusion, we observe significant early-life co-exposure to multiple toxicants, and demonstrate the method's applicability for large-scale exposomics-type cohort studies.
Specific localization and quantification of the Oligo-Mouse-Microbiota (OMM12) by fluorescence in situ hybridization (FISH)
2022 - Current Protocols, 2: e548
Abstract:
The oligo-mouse-microbiota (OMM12) is a widely used syncom that colonizes gnotobiotic mice in a stable manner. It provides several fundamental functions to its murine host, including colonization resistance against enteric pathogens. Here, we designed and validated specific fluorescence in situ hybridization (FISH) probes to detect and quantify OMM12 strains on intestinal tissue cross sections. 16S rRNA‒specific probes were designed, and specificity was validated on fixed pure cultures. A hybridization protocol was optimized for sensitive detection of the individual bacterial cells in cryosections. Using this method, we showed that the intestinal mucosal niche of Akkermansia muciniphila can be influenced by global gut microbial community context.
SRS-FISH: A high-throughput platform linking microbiome metabolism to identity at the single-cell level.
2022 - Proc Natl Acad Sci U S A, 26: e2203519119
Abstract:
One of the biggest challenges in microbiome research in environmental and medical samples is to better understand functional properties of microbial community members at a single-cell level. Single-cell isotope probing has become a key tool for this purpose, but the current detection methods for determination of isotope incorporation into single cells do not allow high-throughput analyses. Here, we report on the development of an imaging-based approach termed stimulated Raman scattering-two-photon fluorescence in situ hybridization (SRS-FISH) for high-throughput metabolism and identity analyses of microbial communities with single-cell resolution. SRS-FISH offers an imaging speed of 10 to 100 ms per cell, which is two to three orders of magnitude faster than achievable by state-of-the-art methods. Using this technique, we delineated metabolic responses of 30,000 individual cells to various mucosal sugars in the human gut microbiome via incorporation of deuterium from heavy water as an activity marker. Application of SRS-FISH to investigate the utilization of host-derived nutrients by two major human gut microbiome taxa revealed that response to mucosal sugars tends to be dominated by Bacteroidales, with an unexpected finding that Clostridia can outperform Bacteroidales at foraging fucose. With high sensitivity and speed, SRS-FISH will enable researchers to probe the fine-scale temporal, spatial, and individual activity patterns of microbial cells in complex communities with unprecedented detail.
Targeting Gut Bacteria Using Inulin-Conjugated Mesoporous Silica Nanoparticles
2022 - Adv Mater Interfaces, 9: 202102558
Abstract:
To facilitate the creation of novel nanocarrier systems targeting the intestinal microbiome, inulin-conjugated mesoporous silica nanoparticles (MSNs) are described herein for the first time. Surface functionalization is achieved on either hydrophilic or hydrophobic mesoporous nanoparticles using different conjugation methods. The targeting performance of the resulting materials is assessed and compared upon incubation with human stool. It appears that amide formation is the most favorable coupling method on hydrophilic MSNs to achieve the desired bioconjugate. Remarkably, high affinity of gut bacteria to the conjugated particles can be obtained, paving the way to novel targeted drug delivery systems.
Ecological Processes Shaping Microbiomes of Extremely Low Birthweight Infants.
2022 - Front Microbiol, 812136
Abstract:
The human microbiome has been implicated in affecting health outcomes in premature infants, but the ecological processes governing early life microbiome assembly remain poorly understood. Here, we investigated microbial community assembly and dynamics in extremely low birth weight infants (ELBWI) over the first 2 weeks of life. We profiled the gut, oral cavity and skin microbiomes over time using 16S rRNA gene amplicon sequencing and evaluated the ecological forces shaping these microbiomes. Though microbiomes at all three body sites were characterized by compositional instability over time and had low body-site specificity (PERMANOVA, = 0.09, = 0.001), they could nonetheless be clustered into four discrete community states. Despite the volatility of these communities, deterministic assembly processes were detectable in this period of initial microbial colonization. To further explore these deterministic dynamics, we developed a probabilistic approach in which we modeled microbiome state transitions in each ELBWI as a Markov process, or a "memoryless" shift, from one community state to another. This analysis revealed that microbiomes from different body sites had distinctive dynamics as well as characteristic equilibrium frequencies. Time-resolved microbiome sampling of premature infants may help to refine and inform clinical practices. Additionally, this work provides an analysis framework for microbial community dynamics based on Markov modeling that can facilitate new insights, not only into neonatal microbiomes but also other human-associated or environmental microbiomes.
Elucidating the role of the gut microbiota in the physiological effects of dietary fiber.
2022 - Microbiome, 1: 77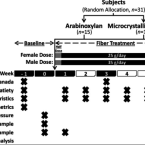
Abstract:
Dietary fiber is an integral part of a healthy diet, but questions remain about the mechanisms that underlie effects and the causal contributions of the gut microbiota. Here, we performed a 6-week exploratory trial in adults with excess weight (BMI: 25-35 kg/m) to compare the effects of a high-dose (females: 25 g/day; males: 35 g/day) supplement of fermentable corn bran arabinoxylan (AX; n = 15) with that of microbiota-non-accessible microcrystalline cellulose (MCC; n = 16). Obesity-related surrogate endpoints and biomarkers of host-microbiome interactions implicated in the pathophysiology of obesity (trimethylamine N-oxide, gut hormones, cytokines, and measures of intestinal barrier integrity) were assessed. We then determined whether clinical outcomes could be predicted by fecal microbiota features or mechanistic biomarkers.
AX enhanced satiety after a meal and decreased homeostatic model assessment of insulin resistance (HOMA-IR), while MCC reduced tumor necrosis factor-α and fecal calprotectin. Machine learning models determined that effects on satiety could be predicted by fecal bacterial taxa that utilized AX, as identified by bioorthogonal non-canonical amino acid tagging. Reductions in HOMA-IR and calprotectin were associated with shifts in fecal bile acids, but correlations were negative, suggesting that the benefits of fiber may not be mediated by their effects on bile acid pools. Biomarkers of host-microbiome interactions often linked to bacterial metabolites derived from fiber fermentation (short-chain fatty acids) were not affected by AX supplementation when compared to non-accessible MCC.
This study demonstrates the efficacy of purified dietary fibers when used as supplements and suggests that satietogenic effects of AX may be linked to bacterial taxa that ferment the fiber or utilize breakdown products. Other effects are likely microbiome independent. The findings provide a basis for fiber-type specific therapeutic applications and their personalization.
Clinicaltrials.gov, NCT02322112 , registered on July 3, 2015. Video Abstract.Individuality of the Extremely Premature Infant Gut Microbiota Is Driven by Ecological Drift.
2022 - mSystems, e0016322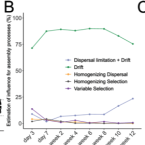
Abstract:
The initial contact between humans and their colonizing gut microbiota after birth is thought to have expansive and long-lasting consequences for physiology and health. Premature infants are at high risk of suffering from lifelong impairments, due in part to aberrant development of gut microbiota that can contribute to early-life infections and inflammation. Despite their importance to health, the ecological assembly and succession processes governing gut microbiome composition in premature infants remained incompletely understood. Here, we quantified these ecological processes in a spatiotemporally resolved 16S rRNA gene amplicon sequencing data set of 60 extremely premature neonates using an established mathematical framework. We found that gut colonization during the first months of life is predominantly stochastic, whereby interindividual diversification of microbiota is driven by ecological drift. Dispersal limitations are initially small but have increasing influence at later stages of succession. Furthermore, we find similar trends in a cohort of 32 healthy term-born infants. These results suggest that the uniqueness of individual gut microbiota of extremely premature infants is largely due to stochastic assembly. Our knowledge concerning the initial gut microbiome assembly in human neonates is limited, and scientific progression in this interdisciplinary field is hindered due to the individuality in composition of gut microbiota. Our study addresses the ecological processes that result in the observed individuality of microbes in the gastrointestinal tract between extremely premature and term-born infants. We find that initial assembly is mainly driven by neutral ecological processes. Interestingly, while this progression is predominantly random, limitations to the dispersal of microbiota between infants become increasingly important with age and are concomitant features of gut microbiome stability. This indicates that while we cannot predict gut microbiota assembly due to its random nature, we can expect the establishment of certain ecological features that are highly relevant for neonatal health.
Differential Modulation of the European Sea Bass Gut Microbiota by Distinct Insect Meals.
2022 - Front Microbiol, 831034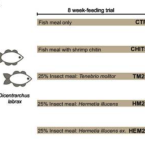
Abstract:
The aquaculture industry is one of the fastest-growing sectors in animal food production. However, farming of carnivorous fish strongly relies on the use of wild fish-based meals, a practice that is environmentally and economically unsustainable. Insect-based diets constitute a strong candidate for fishmeal substitution, due to their high nutritional value and low environmental footprint. Nevertheless, data on the impact of insect meal (IM) on the gut microbiome of farmed fish are so far inconclusive, and very scarce in what concerns modulation of microbial-mediated functions. Here we use high-throughput 16S rRNA gene amplicon sequencing and quantitative PCR to evaluate the impact of different IMs on the composition and chitinolytic potential of the European sea bass gut digesta- and mucosa-associated communities. Our results show that insect-based diets of distinct origins differently impact the gut microbiota of the European sea bass (). We detected clear modulatory effects of IM on the gut microbiota, which were more pronounced in the digesta, where communities differed considerably among the diets tested. Major community shifts were associated with the use of black soldier fly larvae (, HM) and pupal exuviae (HEM) feeds and were characterized by an increase in the relative abundance of the Firmicutes families , , and and the Actinobacteria family , which all include taxa considered beneficial for fish health. Modulation of the digesta community by HEM was characterized by a sharp increase in and a decrease of several Gammaproteobacteria and Bacteroidota members. In turn, a mealworm larvae-based diet (, TM) had only a modest impact on microbiota composition. Further, using quantitative PCR, we demonstrate that shifts induced by HEM were accompanied by an increase in copy number of chitinase ChiA-encoding genes, predominantly originating from species with effective chitinolytic activity. Our study reveals an HEM-driven increase in chitin-degrading taxa and associated chitinolytic activity, uncovering potential benefits of adopting exuviae-supplemented diets, a waste product of insect rearing, as a functional ingredient.
Individual Sweet Taste Perception Influences Salivary Characteristics After Orosensory Stimulation With Sucrose and Noncaloric Sweeteners.
2022 - Front Nutr, 831726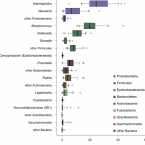
Abstract:
Emerging evidence points to a major role of salivary flow and viscoelastic properties in taste perception and mouthfeel. It has been proposed that sweet-tasting compounds influence salivary characteristics. However, whether perceived differences in the sensory properties of structurally diverse sweet-tasting compounds contribute to salivary flow and saliva viscoelasticity as part of mouthfeel and overall sweet taste perception remains to be clarified. In this study, we hypothesized that the sensory diversity of sweeteners would differentially change salivary characteristics in response to oral sweet taste stimulation. Therefore, we investigated salivary flow and saliva viscoelasticity from 21 healthy test subjects after orosensory stimulation with sucrose, rebaudioside M (RebM), sucralose, and neohesperidin dihydrochalcone (NHDC) in a crossover design and considered the basal level of selected influencing factors, including the basal oral microbiome. All test compounds enhanced the salivary flow rate by up to 1.51 ± 0.12 g/min for RebM compared to 1.10 ± 0.09 g/min for water within the 1st min after stimulation. The increase in flow rate was moderately correlated with the individually perceived sweet taste ( = 0.3, < 0.01) but did not differ between the test compounds. The complex viscosity of saliva was not affected by the test compounds, but the analysis of covariance showed that it was associated ( < 0.05) with mucin 5B (Muc5B) concentration. The oral microbiome was of typical composition and diversity but was strongly individual-dependent (permutational analysis of variance (PERMANOVA): = 0.76, < 0.001) and was not associated with changes in salivary characteristics. In conclusion, this study indicates an impact of individual sweet taste impressions on the flow rate without measurable changes in the complex viscosity of saliva, which may contribute to the overall taste perception and mouthfeel of sweet-tasting compounds.
Persistence of the antagonistic effects of a natural mixture of Alternaria mycotoxins on the estrogen-like activity of human feces after anaerobic incubation.
2022 - Toxicol Lett, 88-99
Abstract:
Several Alternaria mycotoxins are believed to act as endocrine disruptive chemicals (EDCs), since they are reported to bind estrogen receptors in several experimental models. After ingestion of contaminated food commodities, the mycotoxins reach the intestine, where they come into direct contact with food constituents as well as the gut microbiota. Thus, the aim of the present work was to evaluate the modulatory potential of a complex extract of cultured Alternaria fungi (CE; containing eleven chemically characterized compounds) on the estrogenic signaling cascade of mammalian cells before and after anaerobic incubation with fecal slurries, in order to simulate an in vivo-like condition in the gut. Assessing alkaline phosphatase expression in Ishikawa cells as a measure for estrogenicity, we found the CE to partially quench the intrinsic estrogenic properties of fecal slurries and fecal waters, even after 3 h of fecal incubation. Investigation of the mechanisms underlying the effects observed carried out through an in vitro/in silico approach revealed the ability of the extract to decrease the ERα/ERβ nuclear ratio, while a possible action of the mycotoxins as ER-antagonists was excluded. Our results suggest that Alternaria mycotoxins might act as EDCs in vivo, and warrant further investigation in animal models.
Lipid synthesis at the trophic base as the source for energy management to build complex structures.
2022 - Curr Opin Biotechnol, 364-373
Abstract:
The review explores the ecological basis for bacterial lipid metabolism in marine and terrestrial ecosystems. We discuss ecosystem stressors that provoked early organisms to modify their lipid membrane structures, and where these stressors are found across a variety of environments. A major role of lipid membranes is to manage cellular energy utility, including how energy is used for signal propagation. As different environments are imbued with properties that necessitate variation in energy regulation, bacterial lipid synthesis has undergone incalculable permutations of functional trial and error. This may hold clues for how biotechnology can improvise a short-hand version of the evolutionary gauntlet to stimulate latent functional competences for the synthesis of rare lipids. Reducing human reliance on marine resources and deriving solutions for production of essential nutrients is a pressing problem in sustainable agriculture and aquaculture, as well as timely considering the increasing fragility of human health in an aging population.
Raman microspectroscopy for microbiology
2021 - Nature Reviews Methods Primers, 1: 80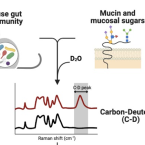
Abstract:
Raman microspectroscopy offers microbiologists a rapid and non-destructive technique to assess the chemical composition of individual live microorganisms in near real time. In this Primer, we outline the methodology and potential for its application to microbiology. We describe the technical aspects of Raman analyses and practical approaches to apply this method to microbiological questions. We discuss recent and potential future applications to determine the composition and distribution of microbial metabolites down to subcellular scale; to investigate the host–microorganism, cell–cell and cell–environment molecular exchanges that underlie the structure of microbial ecosystems from the ocean to the human gut microbiomes; and to interrogate the microbial diversity of functional roles in environmental and industrial processes — key themes in modern microbiology. We describe the current technical limitations of Raman microspectroscopy for investigation of microorganisms and approaches to minimize or address them. Recent technological innovations in Raman microspectroscopy will further reinforce the power and capacity of this method for broader adoptions in microbiology, allowing microbiologists to deepen their understanding of the microbial ecology of complex communities at nearly any scale of interest.
A Mixed-Lipid Emulsion Containing Fish Oil for the Parenteral Nutrition of Preterm Infants: No Impact on Visual Neuronal Conduction.
2021 - Nutrients, 12: in press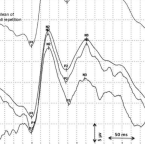
Abstract:
Fish oil is rich in omega-3 fatty acids and essential for neuronal myelination and maturation. The aim of this study was to investigate whether the use of a mixed-lipid emulsion composed of soybean oil, medium-chain triglycerides, olive oil, and fish oil (SMOF-LE) compared to a pure soybean oil-based lipid emulsion (S-LE) for parenteral nutrition had an impact on neuronal conduction in preterm infants. This study is a retrospective matched cohort study comparing preterm infants <1000 g who received SMOF-LE in comparison to S-LE for parenteral nutrition. Visual evoked potentials (VEPs) were assessed longitudinally from birth until discharge. The latencies of the evoked peaks N2 and P2 were analyzed. The analysis included 76 infants (SMOF-LE: = 41 and S-LE: = 35) with 344 VEP measurements (SMOF-LE: = 191 and S-LE = 153). Values of N2 and P2 were not significantly different between the SMOF-LE and S-LE groups. A possible better treatment effect in the SMOF-LE group was seen as a trend toward a shorter latency, indicating faster neural conduction at around term-equivalent age. Prospective trials and follow-up studies are necessary in order to evaluate the potential positive effect of SMOF-LE on neuronal conduction and visual pathway maturation.
Gilbert's Syndrome and the Gut Microbiota - Insights From the Case-Control BILIHEALTH Study.
2021 - Front Cell Infect Microbiol, 701109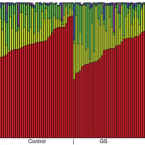
Abstract:
The heme catabolite bilirubin has anti-inflammatory, anti-oxidative and anti-mutagenic effects and its relation to colorectal cancer (CRC) risk is currently under evaluation. Although the main metabolic steps of bilirubin metabolism, including the formation of stercobilin and urobilin, take place in the human gastrointestinal tract, potential interactions with the human gut microbiota are unexplored. This study investigated, whether gut microbiota composition is altered in Gilbert's Syndrome (GS), a mild form of chronically elevated serum unconjugated bilirubin (UCB) compared to matched controls. Potential differences in the incidence of CRC-associated bacterial species in GS were also assessed. To this end, a secondary investigation of the BILIHEALTH study was performed, assessing 45 adults with elevated UCB levels (GS) against 45 age- and sex-matched controls (C). Fecal microbiota analysis was performed using 16S rRNA gene sequencing. No association between mildly increased UCB and the composition of the gut microbiota in this healthy cohort was found. The alpha and beta diversity did not differ between C and GS and both groups showed a typical representation of the known dominant phyla. Furthermore, no difference in abundance of and , which have been associated with the mucosa of CRC patients were observed between the groups. A sequence related to the strain YIT 12065 was identified with a weak association value of 0.521 as an indicator species in the GS group. This strain has been previously associated with a lower body mass index, which is typical for the GS phenotype. Overall, sex was the only driver for an identifiable difference in the study groups, as demonstrated by a greater bacterial diversity in women. After adjusting for confounding factors and multiple testing, we can conclude that the GS phenotype does not affect the composition of the human gut microbiota in this generally healthy study group.
Aberrant gut-microbiota-immune-brain axis development in premature neonates with brain damage.
2021 - Cell Host Microbe, 10: 1558-1572.e6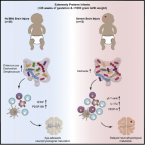
Abstract:
Premature infants are at substantial risk for suffering from perinatal white matter injury. Though the gut microbiota has been implicated in early-life development, a detailed understanding of the gut-microbiota-immune-brain axis in premature neonates is lacking. Here, we profiled the gut microbiota, immunological, and neurophysiological development of 60 extremely premature infants, which received standard hospital care including antibiotics and probiotics. We found that maturation of electrocortical activity is suppressed in infants with severe brain damage. This is accompanied by elevated γδ T cell levels and increased T cell secretion of vascular endothelial growth factor and reduced secretion of neuroprotectants. Notably, Klebsiella overgrowth in the gut is highly predictive for brain damage and is associated with a pro-inflammatory immunological tone. These results suggest that aberrant development of the gut-microbiota-immune-brain axis may drive or exacerbate brain injury in extremely premature neonates and represents a promising target for novel intervention strategies.
In vitro interactions of Alternaria mycotoxins, an emerging class of food contaminants, with the gut microbiota: a bidirectional relationship.
2021 - Arch Toxicol, 7: 2533-2549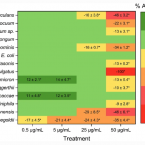
Abstract:
The human gut microbiota plays an important role in the maintenance of human health. Factors able to modify its composition might predispose the host to the development of pathologies. Among the various xenobiotics introduced through the diet, Alternaria mycotoxins are speculated to represent a threat for human health. However, limited data are currently available about the bidirectional relation between gut microbiota and Alternaria mycotoxins. In the present work, we investigated the in vitro effects of different concentrations of a complex extract of Alternaria mycotoxins (CE; containing eleven mycotoxins; e.g. 0.153 µM alternariol and 2.3 µM altersetin, at the maximum CE concentration tested) on human gut bacterial strains, as well as the ability of the latter to metabolize or adsorb these compounds. Results from the minimum inhibitory concentration assay showed the scarce ability of CE to inhibit the growth of the tested strains. However, the growth kinetics of most of the strains were negatively affected by exposure to the various CE concentrations, mainly at the highest dose (50 µg/mL). The CE was also found to antagonize the formation of biofilms, already at concentrations of 0.5 µg/mL. LC-MS/MS data analysis of the mycotoxin concentrations found in bacterial pellets and supernatants after 24 h incubation showed the ability of bacterial strains to adsorb some Alternaria mycotoxins, especially the key toxins alternariol, alternariol monomethyl ether, and altersetin. The tendency of these mycotoxins to accumulate within bacterial pellets, especially in those of Gram-negative strains, was found to be directly related to their lipophilicity.
Anaerobic Sulfur Oxidation Underlies Adaptation of a Chemosynthetic Symbiont to Oxic-Anoxic Interfaces.
2021 - mSystems, 3: e0118620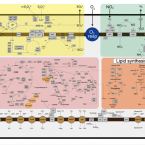
Abstract:
Chemosynthetic symbioses occur worldwide in marine habitats, but comprehensive physiological studies of chemoautotrophic bacteria thriving on animals are scarce. Stilbonematinae are coated by thiotrophic . As these nematodes migrate through the redox zone, their ectosymbionts experience varying oxygen concentrations. However, nothing is known about how these variations affect their physiology. Here, by applying omics, Raman microspectroscopy, and stable isotope labeling, we investigated the effect of oxygen on " Thiosymbion oneisti." Unexpectedly, sulfur oxidation genes were upregulated in anoxic relative to oxic conditions, but carbon fixation genes and incorporation of C-labeled bicarbonate were not. Instead, several genes involved in carbon fixation were upregulated under oxic conditions, together with genes involved in organic carbon assimilation, polyhydroxyalkanoate (PHA) biosynthesis, nitrogen fixation, and urea utilization. Furthermore, in the presence of oxygen, stress-related genes were upregulated together with vitamin biosynthesis genes likely necessary to withstand oxidative stress, and the symbiont appeared to proliferate less. Based on its physiological response to oxygen, we propose that " T. oneisti" may exploit anaerobic sulfur oxidation coupled to denitrification to proliferate in anoxic sand. However, the ectosymbiont would still profit from the oxygen available in superficial sand, as the energy-efficient aerobic respiration would facilitate carbon and nitrogen assimilation. Chemoautotrophic endosymbionts are famous for exploiting sulfur oxidization to feed marine organisms with fixed carbon. However, the physiology of thiotrophic bacteria thriving on the surface of animals (ectosymbionts) is less understood. One longstanding hypothesis posits that attachment to animals that migrate between reduced and oxic environments would boost sulfur oxidation, as the ectosymbionts would alternatively access sulfide and oxygen, the most favorable electron acceptor. Here, we investigated the effect of oxygen on the physiology of " Thiosymbion oneisti," a gammaproteobacterium which lives attached to marine nematodes inhabiting shallow-water sand. Surprisingly, sulfur oxidation genes were upregulated under anoxic relative to oxic conditions. Furthermore, under anoxia, the ectosymbiont appeared to be less stressed and to proliferate more. We propose that animal-mediated access to oxygen, rather than enhancing sulfur oxidation, would facilitate assimilation of carbon and nitrogen by the ectosymbiont.
Mucosal Biofilms Are an Endoscopic Feature of Irritable Bowel Syndrome and Ulcerative Colitis.
2021 - Gastroenterology, 4: 1245-1256.e20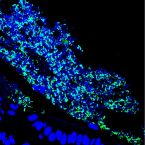
Abstract:
Irritable bowel syndrome (IBS) and inflammatory bowel diseases result in a substantial reduction in quality of life and a considerable socioeconomic impact. In IBS, diagnosis and treatment options are limited, but evidence for involvement of the gut microbiome in disease pathophysiology is emerging. Here we analyzed the prevalence of endoscopically visible mucosal biofilms in gastrointestinal disease and associated changes in microbiome composition and metabolism.
The presence of mucosal biofilms was assessed in 1426 patients at 2 European university-based endoscopy centers. One-hundred and seventeen patients were selected for in-depth molecular and microscopic analysis using 16S ribosomal RNA gene amplicon-sequencing of colonic biopsies and fecal samples, confocal microscopy with deep learning-based image analysis, scanning electron microscopy, metabolomics, and in vitro biofilm formation assays.
Biofilms were present in 57% of patients with IBS and 34% of patients with ulcerative colitis compared with 6% of controls (P < .001). These yellow-green adherent layers of the ileum and right-sided colon were microscopically confirmed to be dense bacterial biofilms. 16S-sequencing links the presence of biofilms to a dysbiotic gut microbiome, including overgrowth of Escherichia coli and Ruminococcus gnavus. R. gnavus isolates cultivated from patient biofilms also formed biofilms in vitro. Metabolomic analysis found an accumulation of bile acids within biofilms that correlated with fecal bile acid excretion, linking this phenotype with a mechanism of diarrhea.
The presence of mucosal biofilms is an endoscopic feature in a subgroup of IBS and ulcerative colitis with disrupted bile acid metabolism and bacterial dysbiosis. They provide novel insight into the pathophysiology of IBS and ulcerative colitis, illustrating that biofilm can be seen as a tipping point in the development of dysbiosis and disease.Combined hormonal contraceptives are associated with minor changes in composition and diversity in gut microbiota of healthy women.
2021 - Environ Microbiol, 6: 3037-3047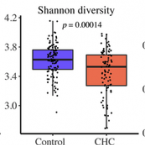
Abstract:
Recent human and animal studies have found associations between gut microbiota composition and serum levels of sex hormones, indicating that they could be an important factor in shaping the microbiota. However, little is known about the effect of regular hormonal fluctuations over the menstrual cycle or CHC-related changes of hormone levels on gut microbiota structure, diversity and dynamics. The aim of this study was to investigate the effect of CHCs on human gut microbiota composition. The effect of CHC pill intake on gut microbiota composition was studied in a group of seven healthy pre-menopausal women using the CHC pill, compared to the control group of nine age-matched healthy women that have not used hormonal contraceptives in the 6 months prior to the start of the study. By analysing the gut microbiota composition in both groups during one menstrual cycle, we found that CHC usage is associated with a minor decrease in gut microbiota diversity and differences in the abundance of several bacterial taxa. These results call for further investigation of the mechanisms underlying hormonal and hormonal contraceptive-related changes of the gut microbiota and the potential implications of these changes for women's health.
Reduced alpha diversity of the oral microbiome correlates with short progression-free survival in patients with relapsed/refractory multiple myeloma treated with ixazomib-based therapy (AGMT MM 1, phase II trial)
2021 - eJHaem, 2: 102-106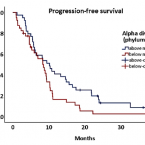
Abstract:
Alterations in the human microbiome have been linked to several malignant diseases. Here, we investigated the oral microbiome of 79 patients with relapsed/refractory multiple myeloma (MM) treated with ixazomib-thalidomide-dexamethasone. Increased alpha diversity (Shannon index) at the phylum level was associated with longer progression-free survival (PFS) (10.2 vs 8.5 months, P = .04), particularly in patients with very long (>75% quartile) PFS . Additionally, alpha diversity was lower in patients with progressive disease (P < .05). These findings suggest an interrelationship between the oral microbiome and outcome in patients with MM and encourage a novel direction for diagnostic and/or therapeutic strategies.
An economical and flexible dual barcoding, two-step PCR approach for highly multiplexed amplicon sequencing
2021 - Front Microbiol, 12: 669776
Abstract:
In microbiome research, phylogenetic and functional marker gene amplicon sequencing is the most commonly-used community profiling approach. Consequently, a plethora of protocols for the preparation and multiplexing of samples for amplicon sequencing have been developed. Here, we present two economical high-throughput gene amplification and sequencing workflows that are implemented as standard operating procedures at the Joint Microbiome Facility of the Medical University of Vienna and the University of Vienna. These workflows are based on a previously-published two-step PCR approach, but have been updated to either increase the accuracy of results, or alternatively to achieve orders of magnitude higher numbers of samples to be multiplexed in a single sequencing run. The high-accuracy workflow relies on unique dual sample barcoding. It allows the same level of sample multiplexing as the previously-published two-step PCR approach, but effectively eliminates residual read missasignments between samples (crosstalk) which are inherent to single barcoding approaches. The high-multiplexing workflow is based on combinatorial dual sample barcoding, which theoretically allows for multiplexing up to 299,756 amplicon libraries of the same target gene in a single massively-parallelized amplicon sequencing run. Both workflows presented here are highly economical, easy to implement, and can, without significant modifications or cost, be applied to any target gene of interest.
Polyphenol Exposure, Metabolism, and Analysis: A Global Exposomics Perspective.
2021 - Annu Rev Food Sci Technol, 461-484
Abstract:
Polyphenols are generally known for their health benefits and estimating actual exposure levels in health-related studies can be improved by human biomonitoring. Here, the application of newly available exposomic and metabolomic technology, notably high-resolution mass spectrometry, in the context of polyphenols and their biotransformation products, is reviewed. Comprehensive workflows for investigating these important bioactives in biological fluids or microbiome-related experiments are scarce. Consequently, this new era of nontargeted analysis and omic-scale exposure assessment offers a unique chance for better assessing exposure to, as well as metabolism of, polyphenols. In clinical and nutritional trials, polyphenols can be investigated simultaneously with the plethora of other chemicals to which we are exposed, i.e., the exposome, which may interact abundantly and modulate bioactivity. This research direction aims at ultimately eluting into atrue systems biology/toxicology evaluation of health effects associated with polyphenol exposure, especially during early life, to unravel their potential for preventing chronic diseases.
Transkingdom interactions between Lactobacilli and hepatic mitochondria attenuate western diet-induced diabetes.
2021 - Nat Commun, 1: 101
Abstract:
Western diet (WD) is one of the major culprits of metabolic disease including type 2 diabetes (T2D) with gut microbiota playing an important role in modulating effects of the diet. Herein, we use a data-driven approach (Transkingdom Network analysis) to model host-microbiome interactions under WD to infer which members of microbiota contribute to the altered host metabolism. Interrogation of this network pointed to taxa with potential beneficial or harmful effects on host's metabolism. We then validate the functional role of the predicted bacteria in regulating metabolism and show that they act via different host pathways. Our gene expression and electron microscopy studies show that two species from Lactobacillus genus act upon mitochondria in the liver leading to the improvement of lipid metabolism. Metabolomics analyses revealed that reduced glutathione may mediate these effects. Our study identifies potential probiotic strains for T2D and provides important insights into mechanisms of their action.
Optofluidic Raman-activated cell sorting for targeted genome retrieval or cultivation of microbial cells with specific functions.
2021 - Nat Protoc, 2: 634-676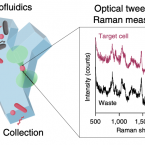
Abstract:
Stable isotope labeling of microbial taxa of interest and their sorting provide an efficient and direct way to answer the question "who does what?" in complex microbial communities when coupled with fluorescence in situ hybridization or downstream 'omics' analyses. We have developed a platform for automated Raman-based sorting in which optical tweezers and microfluidics are used to sort individual cells of interest from microbial communities on the basis of their Raman spectra. This sorting of cells and their downstream DNA analysis, such as by mini-metagenomics or single-cell genomics, or cultivation permits a direct link to be made between the metabolic roles and the genomes of microbial cells within complex microbial communities, as well as targeted isolation of novel microbes with a specific physiology of interest. We describe a protocol from sample preparation through Raman-activated live cell sorting. Subsequent cultivation of sorted cells is described, whereas downstream DNA analysis involves well-established approaches with abundant methods available in the literature. Compared with manual sorting, this technique provides a substantially higher throughput (up to 500 cells per h). Furthermore, the platform has very high sorting accuracy (98.3 ± 1.7%) and is fully automated, thus avoiding user biases that might accompany manual sorting. We anticipate that this protocol will empower in particular environmental and host-associated microbiome research with a versatile tool to elucidate the metabolic contributions of microbial taxa within their complex communities. After a 1-d preparation of cells, sorting takes on the order of 4 h, depending on the number of cells required.
Long-Term Consumption of Anthocyanin-Rich Fruit Juice: Impact on Gut Microbiota and Antioxidant Markers in Lymphocytes of Healthy Males.
2020 - Antioxidants (Basel), 1: in press
Abstract:
Polyphenols are considered protective against diseases associated with oxidative stress. Short-term intake of an anthocyanin-rich fruit juice resulted in significantly reduced deoxyribonucleic acid (DNA) strand-breaks in peripheral blood lymphocytes (PBLs) and affected antioxidant markers in healthy volunteers. Consequently, effects of long-term consumption of fruit juice are of particular interest. In focus was the impact on nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2), the Nrf2-regulated genes NAD(P)H quinone oxidoreductase 1 () and heme oxygenase 1 () as well as effects on the gut microbiota. In a nine-week placebo-controlled intervention trial with 57 healthy male volunteers, consumption of anthocyanin-rich juice significantly increased and transcript levels in PBLs compared to a placebo beverage as measured by real-time polymerase chain reaction (PCR). Three Nrf2-promotor single nucleotide polymorphisms (SNPs), analyzed by pyrosequencing, indicated an association between individual Nrf2 transcript levels and genotype. Moreover, the Nrf2 genotype appeared to correlate with the presence of specific microbial organisms identified by 16S-PCR and classified as . Furthermore, the microbial community was significantly affected by the duration of juice consumption and intake of juice itself. Taken together, long-term consumption of anthocyanin-rich fruit juice affected Nrf2-dependent transcription in PBLs, indicating systemic effects. Individual Nrf2 genotypes may influence the antioxidant response, thus requiring consideration in future intervention studies focusing on the Nrf2 pathway. Anthocyanin-rich fruit juice had an extensive impact on the gut microbiota.
Conversion of Rutin, a Prevalent Dietary Flavonol, by the Human Gut Microbiota.
2020 - Front Microbiol, 585428
Abstract:
The gut microbiota plays a pivotal role in the conversion of dietary flavonoids, which can affect their bioavailability and bioactivity and thereby their health-promoting properties. The ability of flavonoids to metabolically-activate the microbiota has, however, not been systematically evaluated. In the present study, we used a fluorescence-based single-cell activity measure [biorthogonal non-canonical ammino acid-tagging (BONCAT)] combined with fluorescence activated cell sorting (FACS) to determine which microorganisms are metabolically-active after amendment of the flavonoid rutin. We performed anaerobic incubations of human fecal microbiota amended with rutin and in the presence of the cellular activity marker L-azidohomoalanine (AHA) to detect metabolically-active cells. We found that 7.3% of cells in the gut microbiota were active after a 6 h incubation and 26.9% after 24 h. We then sorted BONCAT-positive cells and observed an enrichment of ( and ), , and species in the rutin-responsive fraction of the microbiota. There was marked inter-individual variability in the appearance of rutin conversion products after incubation with rutin. Consistent with this, there was substantial variability in the abundance of rutin-responsive microbiota among different individuals. Specifically, we observed that were associated with conversion of rutin into quercetin-3-glucoside (Q-glc) and were associated with quercetin (Q) production. This suggests that individual microbiotas differ in their ability to metabolize rutin and utilize different conversion pathways.
Transparent soil microcosms for live-cell imaging and non-destructive stable isotope probing of soil microorganisms
2020 - Elife, 9: e56275
Abstract:
Microscale processes are critically important to soil ecology and biogeochemistry yet are difficult to study due to soil’s opacity and complexity. To advance the study of soil processes, we constructed transparent soil microcosms that enable the visualization of microbes via fluorescence microscopy and the non-destructive measurement of microbial activity and carbon uptake in situ via Raman microspectroscopy. We assessed the polymer Nafion and the crystal cryolite as optically transparent soil substrates. We demonstrated that both substrates enable the growth, maintenance, and visualization of microbial cells in three dimensions over time, and are compatible with stable isotope probing using Raman. We applied this system to ascertain that after a dry-down/rewetting cycle, bacteria on and near dead fungal hyphae were more metabolically active than those far from hyphae. These data underscore the impact fungi have facilitating bacterial survival in fluctuating conditions and how these microcosms can yield insights into microscale microbial activities.
Rational design of a microbial consortium of mucosal sugar utilizers reduces Clostridiodes difficile colonization.
2020 - Nat Commun, 1: 5104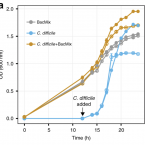
Abstract:
Many intestinal pathogens, including Clostridioides difficile, use mucus-derived sugars as crucial nutrients in the gut. Commensals that compete with pathogens for such nutrients are therefore ecological gatekeepers in healthy guts, and are attractive candidates for therapeutic interventions. Nevertheless, there is a poor understanding of which commensals use mucin-derived sugars in situ as well as their potential to impede pathogen colonization. Here, we identify mouse gut commensals that utilize mucus-derived monosaccharides within complex communities using single-cell stable isotope probing, Raman-activated cell sorting and mini-metagenomics. Sequencing of cell-sorted fractions reveals members of the underexplored family Muribaculaceae as major mucin monosaccharide foragers, followed by members of Lachnospiraceae, Rikenellaceae, and Bacteroidaceae families. Using this information, we assembled a five-member consortium of sialic acid and N-acetylglucosamine utilizers that impedes C. difficile's access to these mucosal sugars and impairs pathogen colonization in antibiotic-treated mice. Our findings underscore the value of targeted approaches to identify organisms utilizing key nutrients and to rationally design effective probiotic mixtures.
Gut microbiota and undigested food constituents modify toxin composition and suppress the genotoxicity of a naturally occurring mixture of Alternaria toxins in vitro.
2020 - Arch Toxicol, 10: 3541-3552
Abstract:
Molds of the genus Alternaria produce several mycotoxins, some of which may pose a threat for health due to their genotoxicity. Due to the lack of adequate toxicological and occurrence data, they are currently not regulated. Interactions between mycotoxins, gut microbiota and food constituents might occur after food ingestion, modifying the bioavailability and, therefore, overall toxicity of mycotoxins. The present work aimed to investigate the impact of in vitro short-term fecal incubation on the in vitro DNA-damaging effects exerted by 5 µg/mL of an Alternaria alternata extract, containing, among others, 15 nM alternariol, 12 nM alternariol monomethyl ether, 241 nM altertoxin II and 301 nM stemphyltoxin III, all of which are known as genotoxic. The involvement of microorganisms, undigested food constituents and soluble substances of human fecal samples in modifying the composition and the genotoxicity of the extract was investigated through the application of LC-MS/MS analysis and comet assays in HT-29 cells. Results showed that the potential of the mycotoxins to induce DNA strand breaks was almost completely quenched, even before anaerobic incubation, by contact with the different fractions of the fecal samples, while the potency to induce formamidopyrimidine DNA glycosylase (FPG)-sensitive sites was only slightly reduced. These effects were in line with a reduction of mycotoxin concentrations found in samples analyzed by LC-MS/MS. Although a direct correlation between the metabolic activity of the gut microbiota and modifications in mycotoxin contents was not clearly observed, adsorptive phenomena to bacterial cells and to undigested food constituents might explain the observed modifications.
Crypt residing bacteria and proximal colonic carcinogenesis in a mouse model of Lynch syndrome.
2020 - Int. J. Cancer, 8: 2316-2326
Abstract:
Colorectal cancer is a multifactorial disease involving inherited DNA mutations, environmental factors, gut inflammation and intestinal microbiota. Certain germline mutations within the DNA mismatch repair system are associated with Lynch syndrome tumors including right-sided colorectal cancer with mucinous phenotype and presence of an inflammatory infiltrate. Such tumors are more often associated with bacterial biofilms, which may contribute to disease onset and progression. Inflammatory bowel diseases are also associated with colorectal cancer and intestinal dysbiosis. Herein we addressed the question, whether inflammation can aggravate colorectal cancer development under mismatch repair deficiency. MSH2 mice were crossed into the IL-10 background to study the importance of inflammation and mucosal bacteria as a driver of tumorigenesis in a Lynch syndrome mouse model. An increase in large bowel tumorigenesis was found in double knockout mice both under conventional housing and under specific pathogen-free conditions. This increase was mostly due to the development of proximal tumors, a hotspot for tumorigenesis in Lynch syndrome, and was associated with a higher degree of inflammation. Additionally, bacterial invasion into the mucus of tumor crypts was observed in the proximal tumors. Inflammation shifted fecal and mucosal microbiota composition and was associated with enrichment in Escherichia-Shigella as well as Akkermansia, Bacteroides and Parabacteroides genera in fecal samples. Tumor-bearing double knockout mice showed a similar enrichment for Escherichia-Shigella and Parabacteroides. Lactobacilli, Lachnospiraceae and Muribaculaceae family members were depleted upon inflammation. In summary, chronic inflammation aggravates colonic tumorigenesis under mismatch repair deficiency and is associated with a shift in microbiota composition.
The role of gut microbiota, butyrate and proton pump inhibitors in amyotrophic lateral sclerosis: a systematic review.
2020 - Int. J. Neurosci., 7: 727-735
Abstract:
We conducted a systematic review on existing literature in humans and animals, linking the gut microbiome with amyotrophic lateral sclerosis (ALS). Additionally, we sought to explore the role of the bacterially produced metabolite butyrate as well as of proton pump inhibitors (PPIs) in these associations. Following PRISMA guidelines for systematic literature reviews, four databases (Medline, Scopus, Embase and Web of Science) were searched and screened by two independent reviewers against defined inclusion criteria. Six studies in humans and six animal studies were identified, summarized and reviewed. Overall, the evidence accrued to date is supportive of changes in the gut microbiome being associated with ALS risk, and potentially progression, though observational studies are small (describing a total of 145 patients with ALS across all published studies), and not entirely conclusive. With emerging studies beginning to apply metagenome sequencing, more clarity regarding the importance and promise of the gut microbiome in ALS can be expected. Future studies may also help establish the therapeutic potential of butyrate, and the role of PPIs in these associations.
Berry-Enriched Diet in Salt-Sensitive Hypertensive Rats: Metabolic Fate of (Poly)Phenols and the Role of Gut Microbiota.
2019 - Nutrients, 11: 2634
Abstract:
Diets rich in (poly)phenols are associated with a reduced reduction in the incidence of cardiovascular disorders. While the absorption and metabolism of (poly)phenols has been described, it is not clear how their metabolic fate is affected under pathological conditions. This study evaluated the metabolic fate of berry (poly)phenols in an in vivo model of hypertension as well as the associated microbiota response. Dahl salt-sensitive rats were fed either a low-salt diet (0.26% NaCl) or a high-salt diet (8% NaCl), with or without a berry mixture (blueberries, blackberries, raspberries, Portuguese crowberry and strawberry tree fruit) for 9 weeks. The salt-enriched diet promoted an increase in the urinary excretion of berry (poly)phenol metabolites, while the abundance of these metabolites decreased in faeces, as revealed by UPLC-MS/MS. Moreover, salt and berries modulated gut microbiota composition as demonstrated by 16S rRNA analysis. Some changes in the microbiota composition were associated with the high-salt diet and revealed an expansion of the families and . However, this effect was mitigated by the dietary supplementation with berries. Alterations in the metabolic fate of (poly)phenols occur in parallel with the modulation of gut microbiota in hypertensive rats. Thus, beneficial effects of (poly)phenols could be related with these interlinked modifications, between metabolites and microbiota environments.
A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome.
2019 - Nat Commun, 1: 4366
Abstract:
Compartmentalization of the gut microbiota is thought to be important to system function, but the extent of spatial organization in the gut ecosystem remains poorly understood. Here, we profile the murine colonic microbiota along longitudinal and lateral axes using laser capture microdissection. We found fine-scale spatial structuring of the microbiota marked by gradients in composition and diversity along the length of the colon. Privation of fiber reduces the diversity of the microbiota and disrupts longitudinal and lateral gradients in microbiota composition. Both mucus-adjacent and luminal communities are influenced by the absence of dietary fiber, with the loss of a characteristic distal colon microbiota and a reduction in the mucosa-adjacent community, concomitant with depletion of the mucus layer. These results indicate that diet has not only global but also local effects on the composition of the gut microbiota, which may affect function and resilience differently depending on location.
Antioxidative activity and health benefits of anthocyanin-rich fruit juice in healthy volunteers.
2019 - Free Radic. Res., 1-11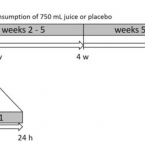
Abstract:
Oxidative cell damage has been linked to the pathogenesis of numerous diseases such as atherosclerosis, type 2 diabetes, and cancer. The consumption of foods rich in polyphenols (e.g. anthocyanins) has been shown to exert preventive effects against such diseases. We investigated the biological effects of anthocyanin-rich fruit juice in a 9-week, placebo-controlled intervention study with 57 healthy male volunteers. The study design encompassed an initial 1 week of wash-out, followed by 8 weeks of intervention period with anthocyanin-rich fruit juice or placebo. The anthocyanin-rich fruit juice demonstrated DNA-protective and antioxidant effects; however, the placebo beverage, rich in vitamin C, showed similar effects based on the tested biomarkers. A significant reduction in background and total DNA strand breaks was observed in both groups within 24 h as well as after 8 weeks of intervention. Only anthocyanin-rich fruit juice consumption provided a significant reduction in body fat and an increase in fat-free mass. The activity of superoxide dismutase (SOD) was significantly elevated after consumption of anthocyanin-rich fruit juice. Both groups showed decreased levels of LDL and total cholesterol (TC) within the first week of the intervention. Similar results in both groups could be explained by the relatively high vitamin C contents of both beverages (>500 mg/L), which may have masked the effects of anthocyanins and other antioxidants in the studied juice. Taken together, anthocyanin-rich fruit juice as well as the placebo drink, both of which had high vitamin C content, can improve DNA integrity and might influence lipid metabolism in humans.
Mucispirillum schaedleri antagonizes Salmonella virulence to protect mice against colitis
2019 - Cell Host Microbe, 25: 681-694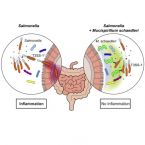
Abstract:
The microbiota and the gastrointestinal mucus layer play a pivotal role in protection against non-typhoidal Salmonellaenterica serovar Typhimurium (S. Tm) colitis. Here, we analyzed the course of Salmonella colitis in mice lacking a functional mucus layer in the gut. Unexpectedly, in contrast to mucus-proficient littermates, genetically deficient mice were protected against Salmonella-induced gut inflammation in the streptomycin colitis model. This correlated with microbiota alterations and enrichment of the bacterial phylum Deferribacteres. Using gnotobiotic mice associated with defined bacterial consortia, we causally linked Mucispirillum schaedleri, currently the sole known representative of Deferribacteres present in the mammalian microbiota, to host protection against S. Tm colitis. Inhibition by M. schaedleri involves interference with S. Tm invasion gene expression, partly by competing for anaerobic electron acceptors. In conclusion, this study establishes M. schaedleri, a core member of the murine gut microbiota, as a key antagonist of S. Tm virulence in the gut.
An automated Raman-based platform for the sorting of live cells by functional properties.
2019 - Nat Microbiol, 6: 1035-1048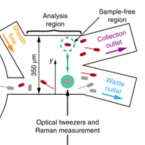
Abstract:
Stable-isotope probing is widely used to study the function of microbial taxa in their natural environment, but sorting of isotopically labelled microbial cells from complex samples for subsequent genomic analysis or cultivation is still in its early infancy. Here, we introduce an optofluidic platform for automated sorting of stable-isotope-probing-labelled microbial cells, combining microfluidics, optical tweezing and Raman microspectroscopy, which yields live cells suitable for subsequent single-cell genomics, mini-metagenomics or cultivation. We describe the design and optimization of this Raman-activated cell-sorting approach, illustrate its operation with four model bacteria (two intestinal, one soil and one marine) and demonstrate its high sorting accuracy (98.3 ± 1.7%), throughput (200-500 cells h; 3.3-8.3 cells min) and compatibility with cultivation. Application of this sorting approach for the metagenomic characterization of bacteria involved in mucin degradation in the mouse colon revealed a diverse consortium of bacteria, including several members of the underexplored family Muribaculaceae, highlighting both the complexity of this niche and the potential of Raman-activated cell sorting for identifying key players in targeted processes.
Microbial nitrogen limitation in the mammalian large intestine.
2018 - Nat Microbiol, 12: 1441-1450
Abstract:
Resource limitation is a fundamental factor governing the composition and function of ecological communities. However, the role of resource supply in structuring the intestinal microbiome has not been established and represents a challenge for mammals that rely on microbial symbionts for digestion: too little supply might starve the microbiome while too much might starve the host. We present evidence that microbiota occupy a habitat that is limited in total nitrogen supply within the large intestines of 30 mammal species. Lowering dietary protein levels in mice reduced their faecal concentrations of bacteria. A gradient of stoichiometry along the length of the gut was consistent with the hypothesis that intestinal nitrogen limitation results from host absorption of dietary nutrients. Nitrogen availability is also likely to be shaped by host-microbe interactions: levels of host-secreted nitrogen were altered in germ-free mice and when bacterial loads were reduced via experimental antibiotic treatment. Single-cell spectrometry revealed that members of the phylum Bacteroidetes consumed nitrogen in the large intestine more readily than other commensal taxa did. Our findings support a model where nitrogen limitation arises from preferential host use of dietary nutrients. We speculate that this resource limitation could enable hosts to regulate microbial communities in the large intestine. Commensal microbiota may have adapted to nitrogen-limited settings, suggesting one reason why excess dietary protein has been associated with degraded gut-microbial ecosystems.
Stable-isotope probing of human and animal microbiome function
2018 - Trends Microbiol, 26: 999-1007
Abstract:
Humans and animals host diverse communities of microorganisms important to their physiology and health. Despite extensive sequencing-based characterization of host-associated microbiomes, there remains a dramatic lack of understanding of microbial functions. Stable-isotope probing (SIP) is a powerful strategy to elucidate the ecophysiology of microorganisms in complex host-associated microbiotas. Here, we suggest that SIP methodologies should be more frequently exploited as part of a holistic functional microbiomics approach. We provide examples of how SIP has been used to study host-associated microbes in vivo and ex vivo. We highlight recent developments in SIP technologies and discuss future directions that will facilitate deeper insights into the function of human and animal microbiomes.
Fluorinated Gold Nanoparticles for Nanostructure Imaging Mass Spectrometry.
2018 - ACS Nano, 7: 6938-6948
Abstract:
Nanostructure imaging mass spectrometry (NIMS) with fluorinated gold nanoparticles (f-AuNPs) is a nanoparticle assisted laser desorption/ionization approach that requires low laser energy and has demonstrated high sensitivity. Here we describe NIMS with f-AuNPs for the comprehensive analysis of metabolites in biological tissues. F-AuNPs assist in desorption/ionization by laser-induced release of the fluorocarbon chains with minimal background noise. Since the energy barrier required to release the fluorocarbons from the AuNPs is minimal, the energy of the laser is maintained in the low μJ/pulse range, thus limiting metabolite in-source fragmentation. Electron microscopy analysis of tissue samples after f-AuNP NIMS shows a distinct "raising" of the surface as compared to matrix assisted laser desorption ionization ablation, indicative of a gentle desorption mechanism aiding in the generation of intact molecular ions. Moreover, the use of perfluorohexane to distribute the f-AuNPs on the tissue creates a hydrophobic environment minimizing metabolite solubilization and spatial dislocation. The transfer of the energy from the incident laser to the analytes through the release of the fluorocarbon chains similarly enhances the desorption/ionization of metabolites of different chemical nature, resulting in heterogeneous metabolome coverage. We performed the approach in a comparative study of the colon of mice exposed to three different diets. F-AuNP NIMS allows the direct detection of carbohydrates, lipids, bile acids, sulfur metabolites, amino acids, nucleotide precursors as well as other small molecules of varied biological origins. Ultimately, the diversified molecular coverage obtained provides a broad picture of a tissue's metabolic organization.
Long-distance electron transport in individual, living cable bacteria.
2018 - Proc. Natl. Acad. Sci. U.S.A., 22: 5786-5791
Abstract:
Electron transport within living cells is essential for energy conservation in all respiring and photosynthetic organisms. While a few bacteria transport electrons over micrometer distances to their surroundings, filaments of cable bacteria are hypothesized to conduct electric currents over centimeter distances. We used resonance Raman microscopy to analyze cytochrome redox states in living cable bacteria. Cable-bacteria filaments were placed in microscope chambers with sulfide as electron source and oxygen as electron sink at opposite ends. Along individual filaments a gradient in cytochrome redox potential was detected, which immediately broke down upon removal of oxygen or laser cutting of the filaments. Without access to oxygen, a rapid shift toward more reduced cytochromes was observed, as electrons were no longer drained from the filament but accumulated in the cellular cytochromes. These results provide direct evidence for long-distance electron transport in living multicellular bacteria.
Vitamin and Amino Acid Auxotrophy in Anaerobic Consortia Operating under Methanogenic Conditions.
2017 - mSystems, 5: e00038-17
Abstract:
Syntrophy among Archaea and Bacteria facilitates the anaerobic degradation of organic compounds to CH4 and CO2. Particularly during aliphatic and aromatic hydrocarbon mineralization, as in the case of crude oil reservoirs and petroleum-contaminated sediments, metabolic interactions between obligate mutualistic microbial partners are of central importance. Using micromanipulation combined with shotgun metagenomic approaches, we describe the genomes of complex consortia within short-chain alkane-degrading cultures operating under methanogenic conditions. Metabolic reconstruction revealed that only a small fraction of genes in the metagenome-assembled genomes encode the capacity for fermentation of alkanes facilitated by energy conservation linked to H2 metabolism. Instead, the presence of inferred lifestyles based on scavenging anabolic products and intermediate fermentation products derived from detrital biomass was a common feature. Additionally, inferred auxotrophy for vitamins and amino acids suggests that the hydrocarbon-degrading microbial assemblages are structured and maintained by multiple interactions beyond the canonical H2-producing and syntrophic alkane degrader-methanogen partnership. Compared to previous work, our report points to a higher order of complexity in microbial consortia engaged in anaerobic hydrocarbon transformation. IMPORTANCE Microbial interactions between Archaea and Bacteria mediate many important chemical transformations in the biosphere from degrading abundant polymers to synthesis of toxic compounds. Two of the most pressing issues in microbial interactions are how consortia are established and how we can modulate these microbial communities to express desirable functions. Here, we propose that public goods (i.e., metabolites of high energy demand in biosynthesis) facilitate energy conservation for life under energy-limited conditions and determine the assembly and function of the consortia. Our report suggests that an understanding of public good dynamics could result in new ways to improve microbial pollutant degradation in anaerobic systems.
Allspice and Clove As Source of Triterpene Acids Activating the G Protein-Coupled Bile Acid Receptor TGR5.
2017 - Front Pharmacol, 8: 468
Abstract:
Worldwide, metabolic diseases such as obesity and type 2 diabetes have reached epidemic proportions. A major regulator of metabolic processes that gained interest in recent years is the bile acid receptor TGR5 (Takeda G protein-coupled receptor 5). This G protein-coupled membrane receptor can be found predominantly in the intestine, where it is mainly responsible for the secretion of the incretins glucagon-like peptide 1 (GLP-1) and peptide YY (PYY). The aim of this study was (i) to identify plant extracts with TGR5-activating potential, (ii) to narrow down their activity to the responsible constituents, and (iii) to assess whether the intestinal microbiota produces transformed metabolites with a different activity profile. Chenodeoxycholic acid (CDCA) served as positive control for both, the applied cell-based luciferase reporter gene assay for TGR5 activity and the biotransformation assay using mouse fecal slurry. The suitability of the workflow was demonstrated by the biotransformation of CDCA to lithocholic acid resulting in a distinct increase in TGR5 activity. Based on a traditional Tibetan formula, 19 plant extracts were selected and investigated for TGR5 activation. Extracts from the commonly used spices Syzygium aromaticum (SaroE, clove), Pimenta dioica (PdioE, allspice), and Kaempferia galanga (KgalE, aromatic ginger) significantly increased TGR5 activity. After biotransformation, only KgalE showed significant differences in its metabolite profile, which, however, did not alter its TGR5 activity compared to non-transformed KgalE. UHPLC-HRMS (high-resolution mass spectrometry) analysis revealed triterpene acids (TTAs) as the main constituents of the extracts SaroE and PdioE. Identification and quantification of TTAs in these two extracts as well as comparison of their TGR5 activity with reconstituted TTA mixtures allowed the attribution of the TGR5 activity to TTAs. EC50s were determined for the main TTAs, i.e., oleanolic acid (2.2 ± 1.6 μM), ursolic acid (1.1 ± 0.2 μM), as well as for the hitherto unknown TGR5 activators corosolic acid (0.5 ± 1.0 μM) and maslinic acid (3.7 ± 0.7 μM). In conclusion, extracts of clove, allspice, and aromatic ginger activate TGR5, which might play a pivotal role in their therapeutic use for the treatment of metabolic diseases. Moreover, the TGR5 activation of SaroE and PdioE could be pinpointed solely to TTAs.
Bottled aqua incognita: Microbiota assembly and dissolved organic matter diversity in natural mineral waters
2017 - Microbiome, 5: 126
Abstract:
Background: Non-carbonated natural mineral waters contain microorganisms that regularly grow after bottling despite low concentrations of dissolved organic matter (DOM). Yet, the compositions of bottled water microbiota and organic substrates that fuel microbial activity, and how both change after bottling, are still largely unknown.
Results: We performed a multifaceted analysis of microbiota and DOM diversity in twelve natural mineral waters from six European countries. 16S rRNA gene-based analyses showed that less than ten species-level operational taxonomic units (OTUs) dominated the bacterial communities in the water phase and associated with the bottle wall after a short phase of post-bottling growth. Members of the betaproteobacterial genera Curvibacter, Aquabacterium, and Polaromonas (Comamonadaceae) grew in most waters and represent ubiquitous, mesophilic, heterotrophic aerobes in bottled waters. Ultrahigh-resolution mass spectrometry of DOM in bottled waters and their corresponding source waters identified thousands of molecular formulae characteristic of mostly refractory, soil-derived DOM.
Conclusions. The bottle environment, including source water physicochemistry, selected for growth of a similar low-diversity microbiota across various bottled waters. Relative abundance changes of hundreds of multi-carbon molecules were related to growth of less than ten abundant OTUs. We thus speculate that individual bacteria cope with oligotrophic conditions by simultaneously consuming diverse DOM molecules.
Evaluating the Detection of Hydrocarbon-Degrading Bacteria in 16S rRNA Gene Sequencing Surveys.
2017 - Front Microbiol, 8: 2460
Abstract:
Hydrocarbonoclastic bacteria (HCB) play a key role in the biodegradation of oil hydrocarbons in marine and other environments. A small number of taxa have been identified as obligate HCB, notably the Gammaproteobacterial genera Alcanivorax, Cycloclasticus, Marinobacter, Neptumonas, Oleiphilus, Oleispira, and Thalassolituus, as well as the Alphaproteobacterial genus Thalassospira. Detection of HCB in amplicon-based sequencing surveys relies on high coverage by PCR primers and accurate taxonomic classification. In this study, we performed a phylogenetic analysis to identify 16S rRNA gene sequence regions that represent the breadth of sequence diversity within these taxa. Using validated sequences, we evaluated 449 universal 16S rRNA gene-targeted bacterial PCR primer pairs for their coverage of these taxa. The results of this analysis provide a practical framework for selection of suitable primer sets for optimal detection of HCB in sequencing surveys.
Vibrational Spectroscopy for Imaging Single Microbial Cells in Complex Biological Samples.
2017 - Front Microbiol, 8: 675
Abstract:
Vibrational spectroscopy is increasingly used for the rapid and non-destructive imaging of environmental and medical samples. Both Raman and Fourier-transform infrared (FT-IR) imaging have been applied to obtain detailed information on the chemical composition of biological materials, ranging from single microbial cells to tissues. Due to its compatibility with methods such as stable isotope labeling for the monitoring of cellular activities, vibrational spectroscopy also holds considerable power as a tool in microbial ecology. Chemical imaging of undisturbed biological systems (such as live cells in their native habitats) presents unique challenges due to the physical and chemical complexity of the samples, potential for spectral interference, and frequent need for real-time measurements. This Mini Review provides a critical synthesis of recent applications of Raman and FT-IR spectroscopy for characterizing complex biological samples, with a focus on developments in single-cell imaging. We also discuss how new spectroscopic methods could be used to overcome current limitations of single-cell analyses. Given the inherent complementarity of Raman and FT-IR spectroscopic methods, we discuss how combining these approaches could enable us to obtain new insights into biological activities either in situ or under conditions that simulate selected properties of the natural environment.
Members of the Oral Microbiota Are Associated with IL-8 Release by Gingival Epithelial Cells in Healthy Individuals.
2017 - Front Microbiol, 8: 416
Abstract:
The triggers for the onset of oral diseases are still poorly understood. The aim of this study was to characterize the oral bacterial community in healthy humans and its association with nutrition, oral hygiene habits, and the release of the inflammatory marker IL-8 from gingival epithelial cells (GECs) with and without stimulation by bacterial endotoxins to identify possible indicator operational taxonomic units (OTUs) associated with inflammatory marker status. GECs from 21 healthy participants (13 females, 8 males) were incubated with or without addition of bacterial lipopolysaccharides (LPSs), and the oral microbiota was profiled using 16S rRNA gene-targeted sequencing. The basal IL-8 release after 6 h was between 9.9 and 98.2 pg/ml, and bacterial communities were characteristic for healthy oral microbiota. The composition of the oral microbiota was associated with basal IL-8 levels, the intake of meat, tea, white wine, sweets and the use of chewing gum, as well as flossing habits, allergies, gender and body mass index. Additionally, eight OTUs were associated with high basal levels of IL-8 and GEC response to LPS, with high basal levels of IL-8, and 1 with low basal levels of IL8. The identification of indicator bacteria in healthy subjects with high levels of IL-8 release is of importance as they may be promising early warning indicators for the possible onset of oral diseases.
HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis
2017 - Cancer Res., 77: 2424-2438
Abstract:
HuR is an RNA-binding protein implicated in immune homeostasis and various cancers, including colorectal cancer. HuR binding to AU-rich elements within the 3' untranslated region of mRNAs encoding oncogenes, growth factors, and various cytokines leads message stability and translation. In this study, we evaluated HuR as a small-molecule target for preventing colorectal cancer in high-risk groups such as those with familial adenomatosis polyposis (FAP) or inflammatory bowel disease (IBD). In human specimens, levels of cytoplasmic HuR were increased in colonic epithelial cells from patients with IBD, IBD-cancer, FAP-adenoma, and colorectal cancer, but not in patients with IBD-dysplasia. Intraperitoneal injection of the HuR small-molecule inhibitor MS-444 in AOM/DSS mice, a model of IBD and inflammatory colon cancer, augmented DSS-induced weight loss and increased tumor multiplicity, size, and invasiveness. MS-444 treatment also abrogated tumor cell apoptosis and depleted tumor-associated eosinophils, accompanied by a decrease in IL18 and eotaxin-1. In contrast, HuR inhibition in APCMin mice, a model of FAP and colon cancer, diminished the number of small intestinal tumors generated. In this setting, fecal microbiota, evaluated by 16S rRNA gene amplicon sequencing, shifted to a state of reduced bacterial diversity, with an increased representation of Prevotella, Akkermansia, and Lachnospiraceae Taken together, our results indicate that HuR activation is an early event in FAP-adenoma but is not present in IBD-dysplasia. Furthermore, our results offer a preclinical proof of concept for HuR inhibition as an effective means of FAP chemoprevention, with caution advised in the setting of IBD.
A 12-week intervention with nonivamide, a TRPV1 agonist, prevents a dietary-induced body fat gain and increases peripheral serotonin in moderately overweight subjects.
2017 - Mol Nutr Food Res, 5: 1600731
Abstract:
A bolus administration of 0.15 mg nonivamide has previously been demonstrated to reduce energy intake in moderately overweight men. This 12-week intervention investigated whether a daily consumption of nonivamide in a protein-based product formulation promotes a reduction in body weight in healthy overweight subjects and affects outcome measures associated with mechanisms regulating food intake, e.g. plasma concentrations of (an)orexigenic hormones, energy substrates as well as changes in fecal microbiota.
Nineteen overweight subjects were randomly assigned to either a control (C) or a nonivamide (NV) group. Changes in the body composition and plasma concentrations of satiating hormones were determined at fasting and 15, 30, 60, 90, and 120 min after a glucose load. Participants were instructed to consume 0.15 mg nonivamide per day in 450 mL of a milk shake additionally to their habitual diet. After treatment, a group difference in body fat mass change (-0.61 ± 0.36% in NV and +1.36 ± 0.38% in C) and an increase in postprandial plasma serotonin were demonstrated. Plasma metabolome and fecal microbiome read outs were not affected.
A daily intake of 0.15 mg nonivamide helps to support to maintain a healthy body composition.Microbial nutrient niches in the gut.
2017 - Environ. Microbiol., 4: 1366-1378
Abstract:
The composition and function of the mammalian gut microbiota has been the subject of much research in recent years, but the principles underlying the assembly and structure of this complex community remain incompletely understood. Processes that shape the gut microbiota are thought to be mostly niche-driven, with environmental factors such as the composition of available nutrients largely determining whether or not an organism can establish. The concept that the nutrient landscape dictates which organisms can successfully colonize and persist in the gut was first proposed in Rolf Freter's nutrient niche theory. In a situation where nutrients are perfectly mixed and there is balanced microbial growth, Freter postulated that an organism can only survive if it is able to utilize one or a few limiting nutrients more efficiently than its competitors. Recent experimental work indicates, however, that nutrients in the gut vary in space and time. We propose that in such a scenario, Freter's nutrient niche theory must be expanded to account for the co-existence of microorganisms utilizing the same nutrients but in distinct sites or at different times, and that metabolic flexibility and mixed-substrate utilization are common strategies for survival in the face of ever-present nutrient fluctuations.
Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota
2017 - mSystems, 2: e00171-16
Abstract:
Mucispirillum schaedleri is an abundant inhabitant of the intestinal mucus layer of rodents and other animals and has been suggested to be a pathobiont, a commensal that plays a role in disease. In order to gain insights into its lifestyle, we analyzed the genome and transcriptome of M. schaedleri ASF 457 and performed physiological experiments to test traits predicted by its genome. Although described as a mucus inhabitant, M. schaedleri has limited capacity for degrading host-derived mucosal glycans and other complex polysaccharides. Additionally, M. schaedleri reduces nitrate and expresses systems for scavenging oxygen and reactive oxygen species in vivo, which may account for its localization close to the mucosal tissue and expansion during inflammation. Also of note, M. schaedleri harbors a type VI secretion system and putative effector proteins and can modify gene expression in mucosal tissue, suggesting intimate interactions with its host and a possible role in inflammation. The M. schaedleri genome has been shaped by extensive horizontal gene transfer, primarily from intestinal Epsilon- and Deltaproteobacteria, indicating that horizontal gene transfer has played a key role in defining its niche in the gut ecosystem.
Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations
2017 - Environ. Microbiol., 1: 95-105
Abstract:
An altered gut microbiota has been linked to obesity in adulthood, although little is known about childhood obesity. The aim of this study was to characterize the composition of the gut microbiota in obese (n = 42) and normal-weight (n = 36) children aged 6 to 16. Using 16S rRNA gene-targeted sequencing, we evaluated taxa with differential abundance according to age- and sex-normalized body mass index (BMI z-score). Obesity was associated with an altered gut microbiota characterized by elevated levels of Firmicutes and depleted levels of Bacteroidetes. Correlation network analysis revealed that the gut microbiota of obese children also had increased correlation density and clustering of operational taxonomic units (OTUs). Members of the Bacteroidetes were generally better predictors of BMI z-score and obesity than Firmicutes, which was likely due to discordant responses of Firmicutes OTUs. In accordance with these observations, the main metabolites produced by gut bacteria, short chain fatty acids (SCFAs), were higher in obese children, suggesting elevated substrate utilisation. Multiple taxa were correlated with SCFA levels, reinforcing the tight link between the microbiota, SCFAs and obesity. Our results suggest that gut microbiota dysbiosis and elevated fermentation activity may be involved in the etiology of childhood obesity.
Genome-guided design of a novel defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium
2016 - Nature Microbiol, 2: 16215
Abstract:
Protection against enteric infections, also termed colonization resistance, results from mutualistic interactions of the host and its indigenous microbes. The gut microbiota of humans and mice is highly diverse and it is therefore challenging to assign specific properties to its individual members. Here, we have used a collection of murine bacterial strains and a modular design approach to create a minimal bacterial community that, once established in germ-free mice, provided colonization resistance against the human enteric pathogen Salmonella enterica serovar Typhimurium (S. Tm). Initially, a community of 12 strains, termed Oligo-Mouse Microbiota (Oligo-MM12), representing members of the major bacterial phyla in the murine gut, was selected. This community was stable over consecutive mouse generations and provided colonization resistance against S. Tm infection, albeit not to the degree of a conventional complex microbiota. Comparative (meta)genome analyses identified functions represented in a conventional microbiome but absent from the Oligo-MM12. By genome-informed design, we created an improved version of the Oligo-MM community harbouring three facultative anaerobic bacteria from the Mouse Intestinal Bacterial Collection (miBC) that provided conventional-like colonization resistance. In conclusion, we have established a highly versatile experimental system that showed efficacy in an enteric infection model. Thus, in combination with exhaustive bacterial strain collections and systems-based approaches, genomeguided design can be used to generate insights into microbe–microbe and microbe–host interactions for the investigation of ecological and disease-relevant mechanisms in the intestine.
Bacterial nutrient foraging in a mouse model of enteral nutrient deprivation: Insight into the gut origin of sepsis
2016 - Am J Physiol Gastrointest Liver Physiol, 311: G734-G743
Abstract:
Total parenteral nutrition (TPN) leads to a shift in small intestinal microbiota with a characteristic dominance of Proteobacteria. This study examined how metabolomic changes within the small bowel support an altered microbial community in enterally deprived mice.
C57BL/6 mice were given TPN or enteral chow. Metabolomic analysis of jejunal contents was performed by liquid chromatography/mass spectrometry (LC/MS). In some experiments, leucine in TPN was partly substituted with (13)C-leucine. Additionally, jejunal contents from TPN dependent and enterally fed mice were gavaged into germ-free mice to reveal if the TPN phenotype was transferrable.
Small bowel contents of TPN mice maintained an amino acid composition similar to that of the TPN solution. Mass spectrometry analysis of small bowel contents of TPN dependent mice showed increased concentration of (13)C compared to fed mice receiving saline enriched with (13)C-leucine. (13)C-leucine added to the serosal side of Ussing chambers showed rapid permeation across TPN-dependent jejunum, suggesting increased transmucosal passage. Single-cell analysis by fluorescence in situ hybridization (FISH) - NanoSIMS demonstrated uptake of (13)C-leucine by TPN-associated bacteria, with preferential uptake by Enterobacteriaceae. Gavage of small bowel effluent from TPN mice into germ-free, fed mice resulted in a trend toward the pro-inflammatory TPN-phenotype with loss of epithelial barrier function.
TPN-dependence leads to increased permeation of TPN-derived nutrients into the small intestinal lumen, where they are predominately utilized by Enterobacteriaceae. The altered metabolomic composition of the intestinal lumen during TPN promotes dysbiosis.Behavior of platinum(iv) complexes in models of tumor hypoxia: cytotoxicity, compound distribution and accumulation
2016 - Metallomics, 4: 422-33
Abstract:
Hypoxia in solid tumors remains a challenge for conventional cancer therapeutics. As a source for resistance, metastasis development and drug bioprocessing, it influences treatment results and disease outcome. Bioreductive platinum(iv) prodrugs might be advantageous over conventional metal-based therapeutics, as biotransformation in a reductive milieu, such as under hypoxia, is required for drug activation. This study deals with a two-step screening of experimental platinum(iv) prodrugs with different rates of reduction and lipophilicity with the aim of identifying the most appropriate compounds for further investigations. In the first step, the cytotoxicity of all compounds was compared in hypoxic multicellular spheroids and monolayer culture using a set of cancer cell lines with different sensitivities to platinum(ii) compounds. Secondly, two selected compounds were tested in hypoxic xenografts in SCID mouse models in comparison to satraplatin, and, additionally, (LA)-ICP-MS-based accumulation and distribution studies were performed for these compounds in hypoxic spheroids and xenografts. Our findings suggest that, while cellular uptake and cytotoxicity strongly correlate with lipophilicity, cytotoxicity under hypoxia compared to non-hypoxic conditions and antitumor activity of platinum(iv) prodrugs are dependent on their rate of reduction.
Response of the bacterial community associated with a cosmopolitan marine diatom to crude oil shows a preference for the biodegradation of aromatic hydrocarbons.
2016 - Environ. Microbiol., 6: 1817-33
Abstract:
Emerging evidence shows that hydrocarbonoclastic bacteria (HCB) may be commonly found associated with phytoplankton in the ocean, but the ecology of these bacteria and how they respond to crude oil remains poorly understood. Here, we used a natural diatom-bacterial assemblage to investigate the diversity and response of HCB associated with a cosmopolitan marine diatom, Skeletonema costatum, to crude oil. Pyrosequencing analysis and qPCR revealed a dramatic transition in the diatom-associated bacterial community, defined initially by a short-lived bloom of Methylophaga (putative oil degraders) that was subsequently succeeded by distinct groups of HCB (Marinobacter, Polycyclovorans, Arenibacter, Parvibaculum, Roseobacter clade), including putative novel phyla, as well as other groups with previously unqualified oil-degrading potential. Interestingly, these oil-enriched organisms contributed to the apparent and exclusive biodegradation of substituted and non-substituted polycyclic aromatic hydrocarbons (PAHs), thereby suggesting that the HCB community associated with the diatom is tuned to specializing in the degradation of PAHs. Furthermore, the formation of marine oil snow (MOS) in oil-amended incubations was consistent with its formation during the Deepwater Horizon oil spill. This work highlights the phycosphere of phytoplankton as an underexplored biotope in the ocean where HCB may contribute importantly to the biodegradation of hydrocarbon contaminants in marine surface waters.
Activity and community structures of sulfate-reducing microorganisms in polar, temperate and tropical marine sediments
2016 - ISME J, 10: 796–809
Abstract:
Temperature has a fundamental impact on the metabolic rates of microorganisms and strongly influences microbial ecology and biogeochemical cycling in the environment. In this study, we examined the catabolic temperature response of natural communities of sulfate-reducing microorganisms (SRM) in polar, temperate, and tropical marine sediments. In short-term sediment incubation experiments with 35S-sulfate, we demonstrated how the cardinal temperatures for sulfate reduction correlate with mean annual sediment temperatures, indicating specific thermal adaptations of the dominant SRM in each of the investigated ecosystems. The community structure of putative SRM in the sediments, as revealed by pyrosequencing of bacterial 16S rRNA gene amplicons and phylogenetic assignment to known SRM taxa, consistently correlated with in situ temperatures, but not with sediment organic carbon concentrations or C:N ratios of organic matter. Additionally, several species-level SRM phylotypes of the class Deltaproteobacteria tended to co-occur at sites with similar mean annual temperatures, regardless of geographic distance. The observed temperature adaptations of SRM imply that environmental temperature is a major controlling variable for physiological selection and ecological and evolutionary differentiation of microbial communities.
Intestinal microbiota signatures associated with inflammation history in mice experiencing recurring colitis
2015 - Front Microbiol, 6: 1408
Abstract:
Acute colitis causes alterations in the intestinal microbiota, but the microbiota is thought to recover after such events. Extreme microbiota alterations are characteristic of human chronic inflammatory bowel diseases, although alterations reported in different studies are divergent and sometimes even contradictory. To better understand the impact of periodic disturbances on the intestinal microbiota and its compositional difference between acute and relapsing colitis, we investigated the beginnings of recurrent inflammation using the dextran sodium sulfate (DSS) mouse model of chemically induced colitis. Using bacterial 16S rRNA gene-targeted pyrosequencing as well as quantitative fluorescence in situ hybridization, we profiled the intestinal and stool microbiota of mice over the course of three rounds of DSS-induced colitis and recovery. We found that characteristic inflammation-associated microbiota could be detected in recovery-phase mice. Successive inflammation episodes further drove the microbiota into an increasingly altered composition post-inflammation, and signatures of colitis history were detectable in the microbiota more sensitively than by pathology analysis. Bacterial indicators of murine colitis history were identified in intestinal and stool samples, with a high degree of consistency between both sample types. Stool may therefore be a promising non-invasive source of bacterial biomarkers that are highly sensitive to inflammation state and history.
Intestinal epithelial cell tyrosine kinase 2 transduces interleukin-22 signals to protect from acute colitis
2015 - J Immunol., 195: 5011-5024
Abstract:
In the intestinal tract, IL-22 activates signal transducer and activator of transcription 3 (STAT3) to promote intestinal epithelial cell (IEC) homeostasis and tissue healing. The mechanism has remained obscure but we demonstrate that IL-22 acts via tyrosine kinase 2 (Tyk2), a member of the Janus kinase (Jak) family. Using a mouse model for colitis, we show that Tyk2 deficiency is associated with an altered composition of the gut microbiota and exacerbates inflammatory bowel disease (IBD). Colitic Tyk2-/- mice have less phosphorylated STAT3 (pY-STAT3) in colon tissue and their IECs proliferate less efficiently. Tyk2-deficient primary IECs show reduced pY-STAT3 in response to IL-22 stimulation and expression of IL-22-STAT3 target genes is reduced in IECs from healthy and colitic Tyk2-/- mice. Experiments with conditional Tyk2-/- mice reveal that IEC-specific depletion of Tyk2 aggravates colitis. Disease symptoms can be alleviated by administering high doses of recombinant IL-22-Fc, indicating that Tyk2 deficiency can be rescued via the IL-22 receptor complex. The pivotal function of Tyk2 in IL-22-dependent colitis was confirmed in Citrobacter rodentium-induced disease. Thus, Tyk2 protects against acute colitis in part by amplifying inflammation-induced epithelial IL-22 signaling to STAT3.
Abstract:
Ammonia- and nitrite-oxidizing microorganisms are collectively responsible for the aerobic oxidation of ammonia via nitrite to nitrate and have essential roles in the global biogeochemical nitrogen cycle. The physiology of nitrifiers has been intensively studied, and urea and ammonia are the only recognized energy sources that promote the aerobic growth of ammonia-oxidizing bacteria and archaea. Here we report the aerobic growth of a pure culture of the ammonia-oxidizing thaumarchaeote Nitrososphaera gargensis using cyanate as the sole source of energy and reductant; to our knowledge, the first organism known to do so. Cyanate, a potentially important source of reduced nitrogen in aquatic and terrestrial ecosystems, is converted to ammonium and carbon dioxide in Nitrososphaera gargensis by a cyanase enzyme that is induced upon addition of this compound. Within the cyanase gene family, this cyanase is a member of a distinct clade also containing cyanases of nitrite-oxidizing bacteria of the genus Nitrospira. We demonstrate by co-culture experiments that these nitrite oxidizers supply cyanase-lacking ammonia oxidizers with ammonium from cyanate, which is fully nitrified by this microbial consortium through reciprocal feeding. By screening a comprehensive set of more than 3,000 publically available metagenomes from environmental samples, we reveal that cyanase-encoding genes clustering with the cyanases of these nitrifiers are widespread in the environment. Our results demonstrate an unexpected metabolic versatility of nitrifying microorganisms, and suggest a previously unrecognized importance of cyanate in cycling of nitrogen compounds in the environment.
A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes.
2015 - Front Microbiol, 6: 731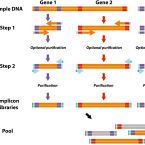
Abstract:
High throughput sequencing of phylogenetic and functional gene amplicons provides tremendous insight into the structure and functional potential of complex microbial communities. Here, we introduce a highly adaptable and economical PCR approach to barcoding and pooling libraries of numerous target genes. In this approach, we replace gene- and sequencing platform-specific fusion primers with general, interchangeable barcoding primers, enabling nearly limitless customized barcode-primer combinations. Compared to barcoding with long fusion primers, our multiple-target gene approach is more economical because it overall requires lower number of primers and is based on short primers with generally lower synthesis and purification costs. To highlight our approach, we pooled over 900 different small-subunit rRNA and functional gene amplicon libraries obtained from various environmental or host-associated microbial community samples into a single, paired-end Illumina MiSeq run. Although the amplicon regions ranged in size from approximately 290 to 720 bp, we found no significant systematic sequencing bias related to amplicon length or gene target. Our results indicate that this flexible multiplexing approach produces large, diverse, and high quality sets of amplicon sequence data for modern studies in microbial ecology.
Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells
2015 - Proc Natl Acad Sci USA, 112: E194-203
Abstract:
Microbial communities are essential to the function of virtually all ecosystems and eukaryotes, including humans. However, it is still a major challenge to identify microbial cells active under natural conditions in complex systems. In this study, we developed a new method to identify and sort active microbes on the single-cell level in complex samples using stable isotope probing with heavy water (D2O) combined with Raman microspectroscopy. Incorporation of D2O-derived D into the biomass of autotrophic and heterotrophic bacteria and archaea could be unambiguously detected via C-D signature peaks in single-cell Raman spectra, and the obtained labeling pattern was confirmed by nanoscale-resolution secondary ion MS. In fast-growing Escherichia coli cells, label detection was already possible after 20 min. For functional analyses of microbial communities, the detection of D incorporation from D2O in individual microbial cells via Raman microspectroscopy can be directly combined with FISH for the identification of active microbes. Applying this approach to mouse cecal microbiota revealed that the host-compound foragers Akkermansia muciniphila and Bacteroides acidifaciens exhibited distinctive response patterns to amendments of mucin and sugars. By Raman-based cell sortingof active (deuterated) cells with optical tweezers and subsequent multiple displacement amplification and DNA sequencing, novel cecal microbes stimulated by mucin and/or glucosamine were identified, demonstrating the potential of the nondestructive D2O-Raman approach for targeted sortingof microbial cells with defined functional properties for single-cell genomics.
Type I interferons have opposing effects during the emergence and recovery phases of colitis.
2014 - Eur J Immunol., 44: 2749-60
Abstract:
The contribution of the innate immune system to inflammatory bowel disease (IBD) is under intensive investigation. Research in animal models has demonstrated that type I interferons (IFN-Is) protect from IBD. In contrast, studies of patients with IBD have produced conflicting results concerning the therapeutic potential of IFN-Is. Here, we present data suggesting that IFN-Is play dual roles as regulators of intestinal inflammation in dextran sodium sulfate (DSS)-treated C57BL/6 mice. Though IFN-Is reduced acute intestinal damage and the abundance ofcolitis-associated intestinal bacteria caused by treatment with a high dose of DSS, they also inhibited the resolution of inflammation after DSS treatment. IFN-Is played an anti-inflammatory role by suppressing the release of IL-1β from the colon MHC class II(+) cells. Consistently, IL-1 receptor blockade reduced the severity of inflammation in IFN-I receptor-deficient mice and myeloid cell-restricted ablation of the IFN-I receptor was detrimental. The proinflammatory role of IFN-Is during recovery from DSS treatment was caused by IFN-I-dependent cell apoptosis as well as an increase in chemokine production and infiltrating inflammatory monocytes and neutrophils. Thus, IFN-Is play opposing roles in specificphases of intestinal injury and inflammation, which may be important for guiding treatment strategies in patients.
Removal of pharmaceuticals and personal care products during water recycling: microbial community structure and effects of substrate concentration.
2014 - Appl Environ Microbiol., 80: 2440-50
Abstract:
Many pharmaceuticals and personal care products (PPCPs) have been shown to be biotransformed in water treatment systems. However, little research exists on the effect of initial PPCP concentration on PPCP biotransformation or on the microbial communities treating impacted water. In this study, biological PPCP removal at various concentrations was assessed using laboratory columns inoculated with wastewater treatment plant effluent. Pyrosequencing was used to examine microbial communities in the columns and in soil from a soil aquifer treatment (SAT; a method ofwater treatment prior to reuse) site. Laboratory columns were supplied with different concentrations (0.25, 10, 100, or 1,000 μg liter(-1)) of each of 15 PPCPs. Five PPCPs (4-isopropyl-3-methylphenol [biosol], p-chloro-m-xylenol, gemfibrozil, ketoprofen, and phenytoin) were not removed at any tested concentrations. Two PPCPs (naproxen and triclosan) exhibited removals independent of PPCP concentration. PPCP removal efficiencies were dependent on initial concentrations for biphenylol, p-chloro-m-cresol, chlorophene, diclofenac, 5-fluorouracil, ibuprofen, and valproic acid, showing that PPCP concentration can affect biotransformation. Biofilms from sand samples collected from the 0.25- and 10-μg liter(-1) PPCP columns were pyrosequenced along with SAT soil samples collected on three consecutive days of a wetting and drying cycle to enable comparison of these two communities exposed to PPCPs. SAT communities were similar to column communities in taxonomy and phylotype composition, and both were found to contain close relatives of known PPCP degraders. The efficiency of biological removal of PPCPs was found to be dependent on the concentration at which the contamination occurs for some, but not all, PPCPs.
Endospores of thermophilic bacteria as tracers of microbial dispersal by ocean currents
2014 - ISME J., 8: 1153-65
Abstract:
Microbial biogeography is influenced by the combined effects of passive dispersal and environmental selection, but the contribution of either factor can be difficult to discern. As thermophilic bacteria cannot grow in the cold seabed, their inactive spores are not subject to environmental selection. We therefore conducted a global experimental survey using thermophilic endospores that are passively deposited by sedimentation to the cold seafloor as tracers to study the effect of dispersal by ocean currents on the biogeography of marine microorganisms. Our analysis of 81 different marine sediments from around the world identified 146 species-level 16S rRNA phylotypes of endospore-forming, thermophilic Firmicutes. Phylotypes showed various patterns of spatial distribution in the world oceans and were dispersal-limited to different degrees. Co-occurrence of several phylotypes in locations separated by great distances (west of Svalbard, the Baltic Sea and the Gulf of California) demonstrated a widespread but not ubiquitous distribution. In contrast, Arctic regions with water masses that are relatively isolated from global ocean circulation (Baffin Bay and east of Svalbard) were characterized by low phylotype richness and different compositions of phylotypes. The observed distribution pattern ofthermophilic endospores in marine sediments suggests that the impact of passive dispersal on marine microbial biogeography is controlled by the connectivity of local water masses to ocean circulation.
Intestinal microbiota reduces genotoxic endpoints induced by high-energy protons.
2014 - Radiat Res, 181: 45-53
Abstract:
Ionizing space radiation causes oxidative DNA damage and triggers oxidative stress responses, and compromised DNA repair mechanisms can lead to increased risk of carcinogenesis. Young adult mice with developed innate and adaptive immune systems that harbored either a conventionalintestinal microbiota (CM) or an intestinal microbiota with a restricted microbial composition (RM) were irradiated with a total dose of 1 Gy delivered by high-energy protons (2.5 GeV/n, LET = 0.2-2 keV/μm) or silicon or iron ions (850 MeV/n, LET ≈ 50 keV/μm and 1 GeV/n, LET = 150 keV/μm, respectively). Six hours after whole-body irradiation, acute chromosomal DNA lesions were observed for RM mice but not CM mice. High-throughput rRNA gene sequencing of intestinal mucosal bacteria showed that Barnesiella intestinihominis and unclassified Bacterodiales were significantly more abundant in male RM mice than CM mice, and phylotype densities changed in irradiated mice. In addition, Helicobacter hepaticus and Bacteroides stercoris were higher in CM than RM mice. Elevated levels of persistently phosphorylated γ-H2AX were observed in RM mice exposed to high-energy protons compared to nonirradiated RM mice, and they also were associated with a decrease of the antioxidant glutathione in peripheral blood measured at four weeks after irradiation. After radiation exposure, CM mice showed lower levels of γ-H2AX phosphorylation than RM mice and an increase in specific RM-associated phylotypes, indicating a down-regulating force on DNA repair by differentially abundant phylotypes in RM versus a radiation-sensitive complex CM.
Polycyclic aromatic hydrocarbon degradation of a phytoplankton-associated Arenibacter spp. and description of Arenibacter algicola sp. nov., an aromatic hydrocarbon-degrading bacterium
2014 - Appl Environ Microbiol., 80: 618-28
Abstract:
Pyrosequencing of the bacterial community associated with a cosmopolitan marine diatom during enrichment with crude oil revealed severalArenibacter phylotypes, of which one (OTU-202) had become significantly enriched by the oil. Since members of the genus Arenibacter have not been previously shown to degrade hydrocarbons, we attempted to isolate a representative strain of this genus in order to directly investigate itshydrocarbon-degrading potential. Based on 16S rRNA sequencing, one isolate (designated strain TG409(T)) exhibited >99% sequence identity to three type strains of this genus. On the basis of phenotypic and genotypic characteristics, strain TG409(T) represents a novel species in the genusArenibacter, for which the name Arenibacter algicola sp. nov. is proposed. We reveal for the first time that polycyclic aromatic hydrocarbon (PAH)degradation is a shared phenotype among members of this genus, indicating that it could be used as a taxonomic marker for this genus. Kinetic data for PAH mineralization rates showed that naphthalene was preferred to phenanthrene, and its mineralization was significantly enhanced in the presence of glass wool (a surrogate for diatom cell surfaces). During enrichment on hydrocarbons, strain TG409(T) emulsified n-tetradecane and crude oil, and cells were found to be preferentially attached to oil droplets, indicating an ability by the strain to express cell surface amphiphilic substances (biosurfactants or bioemulsifiers) as a possible strategy to increase the bioavailability of hydrocarbons. This work adds to our growing knowledge on the diversity of bacterial genera in the ocean contributing to the degradation of oil contaminants and of hydrocarbon-degrading bacteria found living in association with marine eukaryotic phytoplankton.
High-fat diet alters gut microbiota physiology in mice
2014 - ISME J., 8: 295-308
Abstract:
The intestinal microbiota is known to regulate host energy homeostasis and can be influenced by high-calorie diets. However, changes affecting the ecosystem at the functional level are still not well characterized. We measured shifts in cecal bacterial communities in mice fed a carbohydrate orhigh-fat (HF) diet for 12 weeks at the level of the following: (i) diversity and taxa distribution by high-throughput 16S ribosomal RNA gene sequencing; (ii) bulk and single-cell chemical composition by Fourier-transform infrared- (FT-IR) and Raman micro-spectroscopy and (iii) metaproteome and metabolome via high-resolution mass spectrometry. High-fat diet caused shifts in the diversity of dominant gut bacteria and altered the proportion of Ruminococcaceae (decrease) and Rikenellaceae (increase). FT-IR spectroscopy revealed that the impact of the diet on cecal chemical fingerprints is greater than the impact of microbiota composition. Diet-driven changes in biochemical fingerprints of members of the Bacteroidales and Lachnospiraceae were also observed at the level of single cells, indicating that there were distinct differences in cellular composition of dominant phylotypes under different diets. Metaproteome and metabolome analyses based on the occurrence of 1760 bacterial proteins and 86 annotated metabolites revealed distinct HF diet-specific profiles. Alteration of hormonal and anti-microbial networks, bile acid and bilirubin metabolism and shifts towards amino acid and simple sugars metabolism were observed. We conclude that a HF diet markedly affects the gut bacterial ecosystem at the functional level.
Deciphering microbial interactions and detecting keystone species with co-occurrence networks
2014 - Front Microbiol, 5: 219
Abstract:
Co-occurrence networks produced from microbial survey sequencing data are frequently used to identify interactions between community members. While this approach has potential to reveal ecological processes, it has been insufficiently validated due to the technical limitations inherent in studying complex microbial ecosystems. Here, we simulate multi-species microbial communities with known interaction patterns using generalized Lotka-Volterra dynamics. We then construct co-occurrence networks and evaluate how well networks reveal the underlying interactions and how experimental and ecological parameters can affect network inference and interpretation. We find that co-occurrence networks can recapitulate interaction networks under certain conditions, but that they lose interpretability when the effects of habitat filtering become significant. We demonstrate that networks suffer from local hot spots of spurious correlation in the neighborhood of hub species that engage in many interactions. We also identify topological features associated with keystone species in co-occurrence networks. This study provides a substantiated framework to guide environmental microbiologists in the construction and interpretation of co-occurrence networks from microbial survey datasets.
Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery
2014 - ISME J., 8(5):1101-14
Abstract:
Although alterations in gut microbiota composition during acute colitis have been repeatedly observed, associated functional changes and therecovery from dysbiosis received little attention. In this study, we investigated structure and function of the gut microbiota during acute inflammationand recovery in a dextran sodium sulfate (DSS)-colitis mouse model using metatranscriptomics, bacterial 16S rRNA gene amplicon sequencing and monitoring of selected host markers. Parallel to an increase of host markers of inflammation during acute colitis, we observed relative abundance shifts and alterations in phylotype composition of the dominant bacterial orders Clostridiales and Bacteroidales, and an increase of the low abundant Enterobacteriales, Deferribacterales, Verrucomicrobiales and Erysipelotrichales. During recovery, the microbiota began to resume, but did not reach its original composition until the end of the experiment. Microbial gene expression was more resilient to disturbance, with pre-perturbation-type transcript profiles appearing quickly after acute colitis. The decrease of Clostridiales during inflammation correlated with a reduction of transcripts related to butyrate formation, suggesting a disturbance in host-microbe signalling and mucosal nutrient provision. The impact of acute inflammationon the Clostridiales was also characterized by a significant downregulation of their flagellin-encoding genes. In contrast, the abundance of members of the Bacteroidales increased along with an increase in transcripts related to mucin degradation. We propose that acute inflammation triggered a selective reaction of the immune system against flagella of commensals and temporarily altered murine microbiota composition and functions relevant for the host. Despite changes in specific interactions, the host-microbiota homeostasis revealed a remarkable ability for recovery.
NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira
2014 - Environ Microbiol, 16: 3055-3071
Abstract:
Nitrospira are the most widespread and diverse known nitrite-oxidizing bacteria and key nitrifiers in natural and engineered ecosystems. Nevertheless, their ecophysiology and environmental distribution are understudied due to the recalcitrance of Nitrospira to cultivation and the lack of a molecular functional marker, which would allow the detection of Nitrospira in the environment. Here we introduce nxrB, the gene encoding subunit beta of nitrite oxidoreductase, as a functional and phylogenetic marker for Nitrospira. Phylogenetic trees based on nxrB of Nitrospira were largely congruent to 16S rRNA-based phylogenies. By using new nxrB-selective PCR primers, we obtained almost full-length nxrB sequences from Nitrospira cultures, two activated sludge samples, and several geographically and climatically distinct soils. Amplicon pyrosequencing of nxrB fragments from 16 soils revealed a previously unrecognized diversity of terrestrial Nitrospira with 1,801 detected species-level OTUs (using an inferred species threshold of 95% nxrB identity). Richness estimates ranged from 10 to 946 co-existing Nitrospira species per soil. Comparison to an archaeal amoA dataset obtained from the same soils [Environ. Microbiol. 14: 525-539 (2012)] uncovered that ammonia-oxidizing archaea and Nitrospira communities were highly correlated across the soil samples, possibly indicating shared habitat preferences or specific biological interactions among members of these nitrifier groups.
Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation
2013 - Am. J. Gastroenterol., 108: 1620-1630
Abstract:
OBJECTIVES: Fecal microbiota transplantation (FMT) from healthy donors, which is an effective alternative for treatment of Clostridium difficile-associated disease, is being considered for several disorders such as inflammatory bowel disease, irritable bowel syndrome, and metabolic syndrome. Disease remission upon FMT is thought to be facilitated by an efficient colonization of healthy donor microbiota, but knowledge of the composition and temporal stability of patient microbiota after FMT is lacking.METHODS:Five patients with moderately to severely active ulcerative colitis (Mayo score ≥6) and refractory to standard therapy received FMT via nasojejunal tube and enema. In addition to clinical activity and adverse events, the patients' fecal bacterial communities were monitored at multiple time points for up to 12 weeks using 16S rRNA gene-targeted pyrosequencing. RESULTS:FMT elicited fever and a temporary increase of C-reactive protein. Abundant bacteria from donors established in recipients, but the efficiency and stability of donor microbiota colonization varied greatly. A positive clinical response was observed after 12 weeks in one patient whose microbiota had been effectively augmented by FMT. This augmentation was marked by successive colonization of donor-derived phylotypes including the anti-inflammatory and/or short-chain fatty acid-producing Faecalibacterium prausnitzii, Rosebura faecis, and Bacteroides ovatus. Disease severity (as measured by the Mayo score) was associated with an overrepresentation of Enterobacteriaceae and an underrepresentation of Lachnospiraceae. CONCLUSIONS:This study highlights the value of characterizing temporally resolved microbiota dynamics for a better understanding of FMT efficacy and provides potentially useful diagnostic indicators for monitoring FMT success in the treatment of ulcerative colitis.
Intestinal bacteria modify lymphoma incidence and latency by affecting systemic inflammatory state, oxidative stress, and leucocyte genotoxicity
2013 - Cancer Res., 73: 4222-4232
Abstract:
Ataxia-telangiectasia is a genetic disorder associated with high incidence of B-cell lymphoma. Using an ataxia-telangiectasia mouse model, we compared lymphoma incidence in several isogenic mouse colonies harboring different bacterial communities, finding that intestinal microbiota are a major contributor to disease penetrance and latency, lifespan, molecular oxidative stress, and systemic leukocyte genotoxicity. High-throughput sequence analysis of rRNA genes identified mucosa-associated bacterial phylotypes that were colony-specific. Lactobacillus johnsonii, which was deficient in the more cancer-prone mouse colony, was causally tested for its capacity to confer reduced genotoxicity when restored by short-term oral transfer. This intervention decreased systemic genotoxicity, a response associated with reduced basal leukocytes and the cytokine-mediated inflammatory state, and mechanistically linked to the host cell biology of systemic genotoxicity. Our results suggest that intestinal microbiota are a potentially modifiable trait for translational intervention in individuals at risk for B-cell lymphoma, or for other diseases that are driven by genotoxicity or the molecular response to oxidative stress.
Role of bacterial exopolymers (EPS) in the fate of the oil released during the Deepwater Horizon oil spill
2013 - PLoS One, 8(6):e67717
Abstract:
Halomonas species are recognized for producing exopolysaccharides (EPS) exhibiting amphiphilic properties that allow these macromolecules to interface with hydrophobic substrates, such as hydrocarbons. There remains a paucity of knowledge, however, on the potential of Halomonas EPS to influence the biodegradation of hydrocarbons. In this study, the well-characterized amphiphilic EPS produced by Halomonas species strain TG39 was shown to effectively increase the solubilization of aromatic hydrocarbons and enhance their biodegradation by an indigenous microbial community from oil-contaminated surface waters collected during the active phase of the Deepwater Horizon oil spill. Three Halomonas strains were isolated from the Deepwater Horizon site, all of which produced EPS with excellent emulsifying qualities and shared high (97-100%) 16S rRNA sequence identity with strain TG39 and other EPS-producing Halomonas strains. Analysis of pyrosequence data from surface water samples collected during the spill revealed several distinct Halomonas phylotypes, of which some shared a high sequence identity (≥97%) to the Halomonas isolates. Other bacterial groups comprising members with well-characterized EPS-producing qualities, such as Alteromonas, Colwellia and Pseudoalteromonas, were also found enriched in surface waters, suggesting that the total pool of EPS in the Gulf during the spill may have been supplemented by these organisms. Roller bottle incubations with one of the Halomonas isolates from Deepwater Horizon demonstrated its ability to effectively produce oil aggregates and emulsify the oil. The enrichment of EPS-producing bacteria during the spill coupled with their capacity to produce amphiphilic EPS is likely to have contributed to the ultimate removal of the oil and in the formation of oil aggregates, which were a dominant feature observed in contaminated surface waters.
Colonization resistance and microbial ecophysiology: Using gnotobiotic mouse models and single-cell technology to explore the intestinal jungle
2013 - FEMS Microbiol. Rev., 37: 793-829
Abstract:
The highly diverse intestinal microbiota forms a structured community engaged in constant communication with itself and its host and is characterized by extensive ecological interactions. A key benefit that the microbiota affords its host is its ability to protect against infections in a process termed colonization resistance (CR), which remains insufficiently understood. In this review, we connect basic concepts of CR with new insights from recent years and highlight key technological advances in the field of microbial ecology. We present a selection of statistical and bioinformatics tools used to generate hypotheses about synergistic and antagonistic interactions in microbial ecosystems from metagenomic datasets. We emphasize the importance of experimentally testing these hypotheses and discuss the value of gnotobiotic mouse models for investigating specific aspects related to microbiota-host-pathogen interactions in a well-defined experimental system. We further introduce new developments in the area of single-cell analysis using fluorescence in situ hybridization in combination with metabolic stable isotope labeling technologies for studying the in vivo activities of complex community members. These approaches promise to yield novel insights into the mechanisms of CR and intestinal ecophysiology in general, and give researchers the means to experimentally test hypotheses in vivo at varying levels of biological and ecological complexity.
Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing
2013 - Proc. Natl. Acad. Sci. USA, 110: 4720-4725
Abstract:
The animal and human intestinal mucosa secretes an assortment of compounds to establish a physical barrier between the host tissue and intestinal contents, a separation that is vital for health. Some pathogenic microorganisms as well as members of the commensal intestinal microbiota have been shown to be able to break down these secreted compounds. Our understanding of host-compound degradation by the commensal microbiota has been limited to knowledge about simplified model systems because of the difficulty in studying the complex intestinal ecosystem in vivo. In this study, we introduce an approach that overcomes previous technical limitations and allows us to observe which microbial cells in the intestine use host-derived compounds. We added stable isotope-labeled threonine i.v. to mice and combined fluorescence in situ hybridization with high-resolution secondary ion mass spectrometry imaging to characterize utilization of host proteins by individual bacterial cells. We show that two bacterial species, Bacteroides acidifaciens and Akkermansia muciniphila, are important host-protein foragers in vivo. Using gnotobiotic mice we show that microbiota composition determines the magnitude and pattern of foraging by these organisms, demonstrating that a complex microbiota is necessary in order for this niche to be fully exploited. These results underscore the importance of in vivo studies of intestinal microbiota, and the approach presented in this study will be a powerful tool to address many other key questions in animal and human microbiome research.
Intestinal microbiota: a source of novel biomarkers in inflammatory bowel disease?
2013 - Best Pract. Res. Clin. Gastroenterol., 27: 47-58
Abstract:
The human intestine harbors a complex microbial ecosystem that performs manifold functions important to the nutrition and health of its host. Extensive study has revealed that the composition of the intestinal microbiota is altered in individuals with inflammatory bowel disease (IBD). The IBD associated intestinal microbiota generally has reduced species richness and diversity, lower temporal stability, and disruption of the secreted mucus layer structure. Multiple studies have identified certain bacterial taxa that are enriched or depleted in IBD including Enterobacteriaceae, Ruminococcus gnavus, and Desulfovibrio (enriched) and Faecalibacterium prausnitzii, Lachnospiraceae, and Akkermansia (depleted). Additionally, the relative abundance of some taxa appears to correlate with established markers of disease activity such as Enterobacteriaceae (enriched) and Lachnospiraceae (depleted). Signature shifts in fecal microbial community composition may therefore prove to be valuable as diagnostic biomarkers, particularly for longitudinal monitoring of disease activity and response to treatments.
Hydrocarbon-degrading bacteria enriched by the Deepwater Horizon oil spill identified by cultivation and stable isotope probing
2013 - ISME J., 7: 2091-2104
Abstract:
The massive influx of crude oil into the Gulf of Mexico during the Deepwater Horizon disaster triggered dramatic microbial community shifts in surface oil slick and deep plume waters. Previous work had shown several taxa, notably DWH Oceanospirillales, Cycloclasticus and Colwellia, were found enriched in these waters based on their dominance in conventional clone and pyrosequencing libraries and were thought to have played a significant role in the degradation of the oil. However, this type of community analysis data failed to provide direct evidence on the functional properties, such as hydrocarbon degradation of organisms. Using DNA-based stable-isotope probing (DNA-SIP) with uniformly 13C-labelled hydrocarbons, we identified several aliphatic- (Alcanivorax, Marinobacter) and polycyclic aromatic hydrocarbon (PAH)- (Alteromonas, Cycloclasticus, Colwellia) degrading bacteria. We also isolated several strains (Alcanivorax, Alteromonas, Cycloclasticus, Halomonas, Marinobacter and Pseudoalteromonas) with demonstrable hydrocarbon-degrading qualities from surface slick and plume water samples collected during the active phase of the spill. Some of these organisms accounted for the majority of sequence reads representing their respective taxa in a pyrosequencing dataset constructed from the same and additional water column samples. Hitherto, Alcanivorax was not identified in any of the previous water column studies analysing the microbial response to the spill and we discuss its failure to respond to the oil. Collectively, our data provides unequivocal evidence on the hydrocarbon-degrading qualities for some of the dominant taxa enriched in surface and plume waters during the Deepwater Horizon oil spill, and a more complete understanding of their role in the fate of the oil.
A dynamic and complex monochloramine stress response in Escherichia coli revealed by transcriptome analysis
2013 - Water Res., 47: 4978-4985
Abstract:
Despite the widespread use of monochloramine in drinking water treatment, there is surprisingly little information about its mode of action. In this study, DNA microarrays were used to investigate the global gene expression of Escherichia coli cells exposed to sub-lethal concentrations of monochloramine, with a focus on temporal dynamics. Genes induced by monochloramine were associated with several stress response functions, including oxidative stress, DNA repair, multidrug efflux, biofilm formation, antibiotic resistance, and cell wall repair. The diversity of functional associations supports a model of monochloramine action involving multiple cellular targets including cell membranes, nucleic acids, and proteins. These data suggest that E. coli responds to monochloramine exposure by activating diverse defense responses rather than a single antioxidant system and the exposure may also induce biofilm formation. The induction of multidrug efflux pumps and specific antibiotic resistance genes further suggests that exposure to monochloramine may contribute to reduced susceptibility to some antibiotics.
Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis
2012 - ISME J., 6: 2091-106
Abstract:
Human inflammatory bowel disease and experimental colitis models in mice are associated with shifts in intestinal microbiota composition, but it is unclear at what taxonomic/phylogenetic level such microbiota dynamics can be indicative for health or disease. Here, we report that dextran sodium sulfate (DSS)-induced colitis is accompanied by major shifts in the composition and function of the intestinal microbiota of STAT1(-/-) and wild-type mice, as determined by 454 pyrosequencing of bacterial 16S rRNA (gene) amplicons, metatranscriptomics and quantitative fluorescence in situ hybridization of selected phylotypes. The bacterial families Ruminococcaceae, Bacteroidaceae, Enterobacteriaceae, Deferribacteraceae and Verrucomicrobiaceae increased in relative abundance in DSS-treated mice. Comparative 16S rRNA sequence analysis at maximum possible phylogenetic resolution identified several indicator phylotypes for DSS treatment, including the putative mucin degraders Akkermansia and Mucispirillum. The analysis additionally revealed strongly contrasting abundance changes among phylotypes of the same family, particularly within the Lachnospiraceae. These extensive phylotype-level dynamics were hidden when reads were grouped at higher taxonomic levels. Metatranscriptomic analysis provided insights into functional shifts in the murine intestinal microbiota, with increased transcription of genes associated with regulation and cell signaling, carbohydrate metabolism and respiration and decreased transcription of flagellin genes during inflammation. These findings (i) establish the first in-depth inventory of the mouse gut microbiota and its metatranscriptome in the DSS colitis model, (ii) reveal that family-level microbial community analyses are insufficient to reveal important colitis-associated microbiota shifts and (iii) support a scenario of shifting intra-family structure and function in the phylotype-rich and phylogenetically diverse Lachnospiraceae in DSS-treated mice.
Barcoded primers used in multiplex amplicon pyrosequencing bias amplification
2011 - Appl. Environ. Microbiol., 77: 7846-7849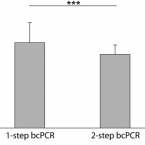
Abstract:
"Barcode-tagged" PCR primers used for multiplex amplicon sequencing generate a thus-far-overlooked amplification bias that produces variable terminal restriction fragment length polymorphism (T-RFLP) and pyrosequencing data from the same environmental DNA template. We propose a simple two-step PCR approach that increases reproducibility and consistently recovers higher genetic diversity in pyrosequencing libraries.
Systematic spatial bias in DNA microarray hybridization is caused by probe spot position-dependent variability in lateral diffusion
2011 - PLoS One, 6: e23727
Abstract:
The hybridization of nucleic acid targets with surface-immobilized probes is a widely used assay for the parallel detection of multiple targets in medical and biological research. Despite its widespread application, DNA microarray technology still suffers from several biases and lack of reproducibility, stemming in part from an incomplete understanding of the processes governing surface hybridization. In particular, non-random spatial variations within individual microarray hybridizations are often observed, but the mechanisms underpinning this positional bias remain incompletely explained.
METHODOLOGY/PRINCIPAL FINDINGS:
This study identifies and rationalizes a systematic spatial bias in the intensity of surface hybridization, characterized by markedly increased signal intensity of spots located at the boundaries of the spotted areas of the microarray slide. Combining observations from a simplified single-probe block array format with predictions from a mathematical model, the mechanism responsible for this biasis found to be a position-dependent variation in lateral diffusion of target molecules. Numerical simulations reveal a strong influence of microarraywell geometry on the spatial bias.
CONCLUSIONS:
Reciprocal adjustment of the size of the microarray hybridization chamber to the area of surface-bound probes is a simple and effective measure to minimize or eliminate the diffusion-based bias, resulting in increased uniformity and accuracy of quantitative DNA microarrayhybridization.
Mycobacterium avium infections of Acanthamoeba strains: host strain variability, grazing-acquired infections, and altered dynamics of inactivation with monochloramine
2010 - Appl. Environ. Microbiol., 76: 6685-6688
Abstract:
Stable Mycobacterium avium infections of several Acanthamoeba strains were characterized by increased infection resistance of recent environmental isolates and reduced infectivity in the presence of other bacteria. Exposure of M. avium in co-culture with Acanthamoeba castellanii to monochloramine yielded inactivation kinetics strikingly similar to those observed for A. castellanii alone.

Book chapters and other publications
STILLLEBEN with Symbionts
2020 - Performance Research, 25: 83-87
Abstract:
STILLLEBEN. Becoming Symbionts’ proposes to value milk and its microbial constituents as primordial assets and currencies - along with cells, sperm, blood, water, and oxygen. The latest scientific research, which has suggested that there is an intimate unseen interplay between mothers and their babies via the transfer of breast milk and microbes, which actually increases the value of the currency with each exchange. The co-operation of Irini Athanassakis and David Berry is an invitation to perceive the transfer of milk not only as an interaction visible to the naked eye, but also on the microscopic level of cells and bacteria. In order to challenge us with this unseen perspective, Athanassakis encourages us to step forward and take a performative and procreative role in expanding our perception. As we are home to billions of microscopic entities, we continuously cast this part of ourselves into our surroundings, impacting and interacting with everything around us. We leave a microbial trace, a lingering residue of cells on the objects, rooms, and people that we encounter. Breast milk and formula are part of such an exchange process. If we look at the logic of microbial exchange in nursing as ‘giving’ and knowledge about symbionts for a future holo-economy based on co-operation and mutualism for collective survival.
The unexpected versatility of the cellulosome
2017 - Environmental Microbiology, 1: 13-14
Hidden potential: Diet-driven changes in redox level shape the rumen microbiome
2017 - Environmental Microbiology, 1: 19-20
Making It Stick: A Compelling Case for Precision Microbiome Reconstitution
2016 - Cell Host & Microbe, 20: 415-417
Abstract:
Modification of the intestinal microbiome is an emerging target to improve health and prevent or treat a number of diseases. In this issue of Cell Host & Microbe, Maldonado-Gomez et al. (2016) uncover the basic principles that govern the successful establishment and persistence of an exogenously introduced gut bacterium.




Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at