Publications
Publications in peer reviewed journals
Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
2024 - Microbiome, 1: 55
Abstract:
Microorganisms are responsible for nutrient removal and resource recovery in wastewater treatment plants (WWTPs), and their diversity is often studied by 16S rRNA gene amplicon sequencing. However, this approach underestimates the abundance and diversity of Patescibacteria due to the low coverage of commonly used PCR primers for this highly divergent bacterial phylum. Therefore, our current understanding of the global diversity, distribution, and ecological role of Patescibacteria in WWTPs is very incomplete. This is particularly relevant as Patescibacteria are considered to be associated with microbial host cells and can therefore influence the abundance and temporal variability of other microbial groups that are important for WWTP functioning.
Here, we evaluated the in silico coverage of widely used 16S rRNA gene-targeted primer pairs and redesigned a primer pair targeting the V4 region of bacterial and archaeal 16S rRNA genes to expand its coverage for Patescibacteria. We then experimentally evaluated and compared the performance of the original and modified V4-targeted primers on 565 WWTP samples from the MiDAS global sample collection. Using the modified primer pair, the percentage of ASVs classified as Patescibacteria increased from 5.9 to 23.8%, and the number of detected patescibacterial genera increased from 560 to 1576, while the detected diversity of the remaining microbial community remained similar. Due to this significantly improved coverage of Patescibacteria, we identified 23 core genera of Patescibacteria in WWTPs and described the global distribution pattern of these unusual microbes in these systems. Finally, correlation network analysis revealed potential host organisms that might be associated with Patescibacteria in WWTPs. Interestingly, strong indications were found for an association between Patescibacteria of the Saccharimonadia and globally abundant polyphosphate-accumulating organisms of the genus Ca. Phosphoribacter.
Our study (i) provides an improved 16S rRNA gene V4 region-targeted amplicon primer pair inclusive of Patescibacteria with little impact on the detection of other taxa, (ii) reveals the diversity and distribution patterns of Patescibacteria in WWTPs on a global scale, and (iii) provides new insights into the ecological role and potential hosts of Patescibacteria in WWTPs. Video Abstract.Taurine as a key intermediate for host-symbiont interaction in the tropical sponge Ianthella basta.
2023 - ISME J, 8: 1208-1223
Abstract:
Marine sponges are critical components of marine benthic fauna assemblages, where their filter-feeding and reef-building capabilities provide bentho-pelagic coupling and crucial habitat. As potentially the oldest representation of a metazoan-microbe symbiosis, they also harbor dense, diverse, and species-specific communities of microbes, which are increasingly recognized for their contributions to dissolved organic matter (DOM) processing. Recent omics-based studies of marine sponge microbiomes have proposed numerous pathways of dissolved metabolite exchange between the host and symbionts within the context of the surrounding environment, but few studies have sought to experimentally interrogate these pathways. By using a combination of metaproteogenomics and laboratory incubations coupled with isotope-based functional assays, we showed that the dominant gammaproteobacterial symbiont, 'Candidatus Taurinisymbion ianthellae', residing in the marine sponge, Ianthella basta, expresses a pathway for the import and dissimilation of taurine, a ubiquitously occurring sulfonate metabolite in marine sponges. 'Candidatus Taurinisymbion ianthellae' incorporates taurine-derived carbon and nitrogen while, at the same time, oxidizing the dissimilated sulfite into sulfate for export. Furthermore, we found that taurine-derived ammonia is exported by the symbiont for immediate oxidation by the dominant ammonia-oxidizing thaumarchaeal symbiont, 'Candidatus Nitrosospongia ianthellae'. Metaproteogenomic analyses also suggest that 'Candidatus Taurinisymbion ianthellae' imports DMSP and possesses both pathways for DMSP demethylation and cleavage, enabling it to use this compound as a carbon and sulfur source for biomass, as well as for energy conservation. These results highlight the important role of biogenic sulfur compounds in the interplay between Ianthella basta and its microbial symbionts.
Cable bacteria with electric connection to oxygen attract flocks of diverse bacteria.
2023 - Nat Commun, 1: 1614
Abstract:
Cable bacteria are centimeter-long filamentous bacteria that conduct electrons via internal wires, thus coupling sulfide oxidation in deeper, anoxic sediment with oxygen reduction in surface sediment. This activity induces geochemical changes in the sediment, and other bacterial groups appear to benefit from the electrical connection to oxygen. Here, we report that diverse bacteria swim in a tight flock around the anoxic part of oxygen-respiring cable bacteria and disperse immediately when the connection to oxygen is disrupted (by cutting the cable bacteria with a laser). Raman microscopy shows that flocking bacteria are more oxidized when closer to the cable bacteria, but physical contact seems to be rare and brief, which suggests potential transfer of electrons via unidentified soluble intermediates. Metagenomic analysis indicates that most of the flocking bacteria appear to be aerobes, including organotrophs, sulfide oxidizers, and possibly iron oxidizers, which might transfer electrons to cable bacteria for respiration. The association and close interaction with such diverse partners might explain how oxygen via cable bacteria can affect microbial communities and processes far into anoxic environments.
Rapid nitrification involving comammox and canonical Nitrospira at extreme pH in saline-alkaline lakes.
2023 - Environ Microbiol, 25: 1055-1067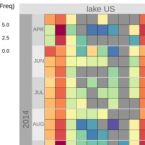
Abstract:
Nitrite-oxidizing bacteria (NOB) catalyse the second nitrification step and are the main biological source of nitrate. The most diverse and widespread NOB genus is Nitrospira, which also contains complete ammonia oxidizers (comammox) that oxidize ammonia to nitrate. To date, little is known about the occurrence and biology of comammox and canonical nitrite oxidizing Nitrospira in extremely alkaline environments. Here, we studied the seasonal distribution and diversity, and the effect of short-term pH changes on comammox and canonical Nitrospira in sediments of two saline, highly alkaline lakes. We identified diverse canonical and comammox Nitrospira clade A-like phylotypes as the only detectable NOB during more than a year, suggesting their major importance for nitrification in these habitats. Gross nitrification rates measured in microcosm incubations were highest at pH 10 and considerably faster than reported for other natural, aquatic environments. Nitrification could be attributed to canonical and comammox Nitrospira and to Nitrososphaerales ammonia-oxidizing archaea. Furthermore, our data suggested that comammox Nitrospira contributed to ammonia oxidation at an extremely alkaline pH of 11. These results identify saline, highly alkaline lake sediments as environments of uniquely strong nitrification with novel comammox Nitrospira as key microbial players.
Mid-Infrared Photothermal-Fluorescence In Situ Hybridization for Functional Analysis and Genetic Identification of Single Cells.
2023 - Anal Chem, 4: 2398-2405
Abstract:
Simultaneous identification and metabolic analysis of microbes with single-cell resolution and high throughput are necessary to answer the question of "who eats what, when, and where" in complex microbial communities. Here, we present a mid-infrared photothermal-fluorescence in situ hybridization (MIP-FISH) platform that enables direct bridging of genotype and phenotype. Through multiple improvements of MIP imaging, the sensitive detection of isotopically labeled compounds incorporated into proteins of individual bacterial cells became possible, while simultaneous detection of FISH labeling with rRNA-targeted probes enabled the identification of the analyzed cells. In proof-of-concept experiments, we showed that the clear spectral red shift in the protein amide I region due to incorporation of C atoms originating from C-labeled glucose can be exploited by MIP-FISH to discriminate and identify C-labeled bacterial cells within a complex human gut microbiome sample. The presented methods open new opportunities for single-cell structure-function analyses for microbiology.
Metabolic reconstruction of the near complete microbiome of the model sponge Ianthella basta.
2023 - Environ Microbiol, 3: 646-660
Abstract:
Many marine sponges host highly diverse microbiomes that contribute to various aspects of host health. Although the putative function of individual groups of sponge symbionts has been increasingly described, the extreme diversity has generally precluded in-depth characterization of entire microbiomes, including identification of syntrophic partnerships. The Indo-Pacific sponge Ianthella basta is emerging as a model organism for symbiosis research, hosting only three dominant symbionts: a Thaumarchaeotum, a Gammaproteobacterium, and an Alphaproteobacterium and a range of other low abundance or transitory taxa. Here, we retrieved metagenome assembled genomes (MAGs) representing >90% of I. basta's microbial community, facilitating the metabolic reconstruction of the sponge's near complete microbiome. Through this analysis, we identified metabolic complementarity between microbes, including vitamin sharing, described the importance of low abundance symbionts, and characterized a novel microbe-host attachment mechanism in the Alphaproteobacterium. We further identified putative viral sequences, highlighting the role viruses can play in maintaining symbioses in I. basta through the horizontal transfer of eukaryotic-like proteins, and complemented this data with metaproteomics to identify active metabolic pathways in bacteria, archaea, and viruses. This data provide the framework to adopt I. basta as a model organism for studying host-microbe interactions and provide a basis for in-depth physiological experiments.
Microbes From Mum: Symbiont transmission in the tropical reef sponge Ianthella basta
2022 - ISME Commun, 2: 90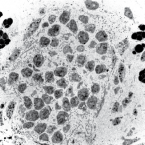
Abstract:
Most marine sponge species harbour distinct communities of microorganisms which contribute to various aspects of their host’s health and physiology. In addition to their key roles in nutrient transformations and chemical defence, these symbiotic microbes may shape sponge phenotype by mediating important developmental stages and influencing the environmental tolerance of the host. However, the characterisation of each microbial taxa throughout a sponge’s life cycle remains challenging, with several sponge species hosting up to 3 000 distinct microbial species. Ianthella basta, an abundant broadcast spawning species in the Indo-Pacific is an emerging model for sponge symbiosis research as it harbours only three dominant symbionts: a Thaumarchaeotum, a Gammaproteobacterium, and an Alphaproteobacterium. Here, we successfully spawned Ianthella basta, characterised its mode of reproduction, and used 16S rRNA gene amplicon sequencing, fluorescence in situ hybridisation, and transmission electron microscopy to characterise the microbial community throughout its life cycle. We confirmed I. basta as being gonochoric and showed that the three dominant symbionts, which together make up >90% of the microbiome according to 16S rRNA gene abundance, are vertically transmitted from mother to offspring by a unique method involving encapsulation in the peri-oocytic space, suggesting an obligate relationship between these microbes and their host.
SRS-FISH: A high-throughput platform linking microbiome metabolism to identity at the single-cell level.
2022 - Proc Natl Acad Sci U S A, 26: e2203519119
Abstract:
One of the biggest challenges in microbiome research in environmental and medical samples is to better understand functional properties of microbial community members at a single-cell level. Single-cell isotope probing has become a key tool for this purpose, but the current detection methods for determination of isotope incorporation into single cells do not allow high-throughput analyses. Here, we report on the development of an imaging-based approach termed stimulated Raman scattering-two-photon fluorescence in situ hybridization (SRS-FISH) for high-throughput metabolism and identity analyses of microbial communities with single-cell resolution. SRS-FISH offers an imaging speed of 10 to 100 ms per cell, which is two to three orders of magnitude faster than achievable by state-of-the-art methods. Using this technique, we delineated metabolic responses of 30,000 individual cells to various mucosal sugars in the human gut microbiome via incorporation of deuterium from heavy water as an activity marker. Application of SRS-FISH to investigate the utilization of host-derived nutrients by two major human gut microbiome taxa revealed that response to mucosal sugars tends to be dominated by Bacteroidales, with an unexpected finding that Clostridia can outperform Bacteroidales at foraging fucose. With high sensitivity and speed, SRS-FISH will enable researchers to probe the fine-scale temporal, spatial, and individual activity patterns of microbial cells in complex communities with unprecedented detail.
The novel genus, 'Candidatus Phosphoribacter', previously identified as Tetrasphaera, is the dominant polyphosphate accumulating lineage in EBPR wastewater treatment plants worldwide.
2022 - ISME J, 6: 1605-1616
Abstract:
The bacterial genus Tetrasphaera encompasses abundant polyphosphate accumulating organisms (PAOs) that are responsible for enhanced biological phosphorus removal (EBPR) in wastewater treatment plants. Recent analyses of genomes from pure cultures revealed that 16S rRNA genes cannot resolve the lineage, and that Tetrasphaera spp. are from several different genera within the Dermatophilaceae. Here, we examine 14 recently recovered high-quality metagenome-assembled genomes from wastewater treatment plants containing full-length 16S rRNA genes identified as Tetrasphaera, 11 of which belong to the uncultured Tetrasphaera clade 3. We find that this clade represents two distinct genera, named here Ca. Phosphoribacter and Ca. Lutibacillus, and reveal that the widely used model organism Tetrasphaera elongata is less relevant for physiological predictions of this uncultured group. Ca. Phosphoribacter incorporates species diversity unresolved at the 16S rRNA gene level, with the two most abundant and often co-occurring species encoding identical V1-V3 16S rRNA gene amplicon sequence variants but different metabolic capabilities, and possibly, niches. Both Ca. P. hodrii and Ca. P. baldrii were visualised using fluorescence in situ hybridisation (FISH), and PAO capabilities were confirmed with FISH-Raman microspectroscopy and phosphate cycling experiments. Ca. Phosphoribacter represents the most abundant former Tetrasphaera lineage and PAO in EPBR systems in Denmark and globally.
Ammonia-oxidizing archaea possess a wide range of cellular ammonia affinities.
2022 - ISME J, 16: 272-283
Abstract:
Nitrification, the oxidation of ammonia to nitrate, is an essential process in the biogeochemical nitrogen cycle. The first step of nitrification, ammonia oxidation, is performed by three, often co-occurring guilds of chemolithoautotrophs: ammonia-oxidizing bacteria (AOB), archaea (AOA), and complete ammonia oxidizers (comammox). Substrate kinetics are considered to be a major niche-differentiating factor between these guilds, but few AOA strains have been kinetically characterized. Here, the ammonia oxidation kinetic properties of 12 AOA representing all major cultivated phylogenetic lineages were determined using microrespirometry. Members of the genus Nitrosocosmicus have the lowest affinity for both ammonia and total ammonium of any characterized AOA, and these values are similar to previously determined ammonia and total ammonium affinities of AOB. This contrasts previous assumptions that all AOA possess much higher substrate affinities than their comammox or AOB counterparts. The substrate affinity of ammonia oxidizers correlated with their cell surface area to volume ratios. In addition, kinetic measurements across a range of pH values supports the hypothesis that-like for AOB-ammonia and not ammonium is the substrate for the ammonia monooxygenase enzyme of AOA and comammox. Together, these data will facilitate predictions and interpretation of ammonia oxidizer community structures and provide a robust basis for establishing testable hypotheses on competition between AOB, AOA, and comammox.
Albumin-targeting of an oxaliplatin-releasing platinum(iv) prodrug results in pronounced anticancer activity due to endocytotic drug uptake .
2021 - Chem Sci, 38: 12587-12599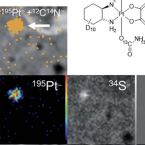
Abstract:
Oxaliplatin is a very potent platinum(ii) drug which is frequently used in poly-chemotherapy schemes against advanced colorectal cancer. However, its benefit is limited by severe adverse effects as well as resistance development. Based on their higher tolerability, platinum(iv) prodrugs came into focus of interest. However, comparable to their platinum(ii) counterparts they lack tumor specificity and are frequently prematurely activated in the blood circulation. With the aim to exploit the enhanced albumin consumption and accumulation in the malignant tissue, we have recently developed a new albumin-targeted prodrug, which supposed to release oxaliplatin in a highly tumor-specific manner. In more detail, we designed a platinum(iv) complex containing two maleimide moieties in the axial position (KP2156), which allows selective binding to the cysteine 34. In the present study, diverse cell biological and analytical tools such as laser ablation inductively-coupled plasma mass spectrometry (LA-ICP-MS), isotope labeling, and nano-scale secondary ion mass spectrometry (NanoSIMS) were employed to better understand the distribution and activation process of KP2156 (in comparison to free oxaliplatin and a non-albumin-binding succinimide analogue). KP2156 forms very stable albumin adducts in the bloodstream resulting in a superior pharmacological profile, such as distinctly prolonged terminal excretion half-life and enhanced effective platinum dose (measured by ICP-MS). The albumin-bound drug is accumulating in the malignant tissue, where it enters the cancer cells clathrin- and caveolin-dependent endocytosis, and is activated by reduction to release oxaliplatin. This results in profound, long-lasting anticancer activity of KP2156 against CT26 colon cancer tumors based on cell cycle arrest and apoptotic cell death. Summarizing, albumin-binding of platinum(iv) complexes potently enhances the efficacy of oxaliplatin therapy and should be further developed towards clinical phase I trials.
Raman microspectroscopy for microbiology
2021 - Nature Reviews Methods Primers, 1: 80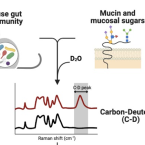
Abstract:
Raman microspectroscopy offers microbiologists a rapid and non-destructive technique to assess the chemical composition of individual live microorganisms in near real time. In this Primer, we outline the methodology and potential for its application to microbiology. We describe the technical aspects of Raman analyses and practical approaches to apply this method to microbiological questions. We discuss recent and potential future applications to determine the composition and distribution of microbial metabolites down to subcellular scale; to investigate the host–microorganism, cell–cell and cell–environment molecular exchanges that underlie the structure of microbial ecosystems from the ocean to the human gut microbiomes; and to interrogate the microbial diversity of functional roles in environmental and industrial processes — key themes in modern microbiology. We describe the current technical limitations of Raman microspectroscopy for investigation of microorganisms and approaches to minimize or address them. Recent technological innovations in Raman microspectroscopy will further reinforce the power and capacity of this method for broader adoptions in microbiology, allowing microbiologists to deepen their understanding of the microbial ecology of complex communities at nearly any scale of interest.
Nitrogen kinetic isotope effects of nitrification by the complete ammonia oxidizer Nitrospira inopinata
2021 - mSphere, 6: e00634-21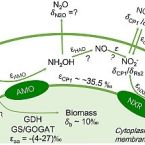
Abstract:
Analysis of nitrogen isotope fractionation effects is useful for tracing biogeochemical nitrogen cycle processes. Nitrification can cause large nitrogen isotope effects through the enzymatic oxidation of ammonia (NH3) via nitrite (NO2-) to nitrate (NO3-) (15εNH4+→NO2- and 15ɛNO2-→NO3-). The isotope effects of ammonia-oxidizing bacteria (AOB) and archaea (AOA), and nitrite-oxidizing bacteria (NOB), have been analyzed previously. Here we studied the nitrogen isotope effects of the complete ammonia oxidizer (comammox) Nitrospira inopinata that oxidizes NH3 to NO3-. At high ammonium (NH4+) availability (1 mM) and pH between 6.5 and 8.5, its 15εNH4+→NO2- ranged from −33.1 to −27.1‰ based on substrate consumption (residual substrate isotopic composition) and −35.5 to −31.2‰ based on product formation (cumulative product isotopic composition), while the 15ɛNO2-→NO3- ranged from 6.5 to 11.1‰ based on substrate consumption. These values resemble isotope effects of AOB and AOA, and of NOB in the genus Nitrospira, suggesting the absence of fundamental mechanistic differences between key enzymes for ammonia and nitrite oxidation in comammox and canonical nitrifiers. However, ambient pH and initial NH4+ concentrations influenced the isotope effects in N. inopinata. The 15εNH4+→NO2- based on product formation was smaller at pH 6.5 (−31.2‰) compared to pH 7.5 (−35.5‰) and pH 8.5 (−34.9‰), while 15ɛNO2-→NO3- was smaller at pH 8.5 (6.5‰) compared to pH 7.5 (8.8‰) and pH 6.5 (11.1‰). Isotopic fractionation via 15εNH4+→NO2- and 15ɛNO2-→NO3- was smaller at 0.1 mM NH4+ compared to 0.5 to 1.0 mM NH4+. Environmental factors, such as pH and NH4+ availability, therefore need to be considered when using isotope effects in 15N isotope fractionation models of nitrification.
Novel Alcaligenes ammonioxydans sp. nov. from wastewater treatment sludge oxidizes ammonia to N with a previously unknown pathway.
2021 - Environ Microbiol, 11: 6965-6980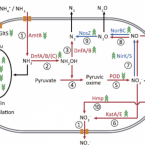
Abstract:
Heterotrophic nitrifiers are able to oxidize and remove ammonia from nitrogen-rich wastewaters but the genetic elements of heterotrophic ammonia oxidation are poorly understood. Here, we isolated and identified a novel heterotrophic nitrifier, Alcaligenes ammonioxydans sp. nov. strain HO-1, oxidizing ammonia to hydroxylamine and ending in the production of N gas. Genome analysis revealed that strain HO-1 encoded a complete denitrification pathway but lacks any genes coding for homologous to known ammonia monooxygenases or hydroxylamine oxidoreductases. Our results demonstrated strain HO-1 denitrified nitrite (not nitrate) to N and N O at anaerobic and aerobic conditions respectively. Further experiments demonstrated that inhibition of aerobic denitrification did not stop ammonia oxidation and N production. A gene cluster (dnfT1RT2ABCD) was cloned from strain HO-1 and enabled E. coli accumulated hydroxylamine. Sub-cloning showed that genetic cluster dnfAB or dnfABC already enabled E. coli cells to produce hydroxylamine and further to N from ( NH ) SO . Transcriptome analysis revealed these three genes dnfA, dnfB and dnfC were significantly upregulated in response to ammonia stimulation. Taken together, we concluded that strain HO-1 has a novel dnf genetic cluster for ammonia oxidation and this dnf genetic cluster encoded a previously unknown pathway of direct ammonia oxidation (Dirammox) to N .
Sensitivity and specificity of the antigen-based anterior nasal self-testing programme for detecting SARS-CoV-2 infection in schools, Austria, March 2021.
2021 - Euro Surveill, 26: pii=2100797
Abstract:
This study evaluates the performance of the antigen-based anterior nasal screening programme implemented in all Austrian schools to detect SARS-CoV-2 infections. We combined nationwide antigen-based screening data obtained in March 2021 from 5,370 schools (Grade 1-8) with an RT-qPCR-based prospective cohort study comprising a representative sample of 244 schools. Considering a range of assumptions, only a subset of infected individuals are detected with the programme (low to moderate sensitivity) and non-infected individuals mainly tested negative (very high specificity).
Cyanate is a low abundance but actively cycled nitrogen compound in soil
2021 - Communications Earth & Environment, 2: 161
Abstract:
Cyanate can serve as a nitrogen and/or carbon source for different microorganisms and as an energy source for autotrophic ammonia oxidizers. However, the extent of cyanate availability and utilisation in terrestrial ecosystems and its role in biogeochemical cycles is poorly known. Here we analyse cyanate concentrations in soils across a range of soil types, land management practices and climates. Soil cyanate concentrations were three orders of magnitude lower than ammonium or nitrate. We determined cyanate consumption in a grassland and rice paddy soil using stable isotope tracer experiments. We find that cyanate turnover was rapid and dominated by biotic processes. We estimated that in-situ cyanate production rates were similar to those associated with urea fertilizer decomposition, a major source of cyanate in the environment. We provide evidence that cyanate is actively turned over in soils and represents a small but continuous nitrogen/energy source for soil microbes.
Anaerobic Sulfur Oxidation Underlies Adaptation of a Chemosynthetic Symbiont to Oxic-Anoxic Interfaces.
2021 - mSystems, 3: e0118620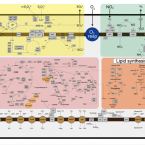
Abstract:
Chemosynthetic symbioses occur worldwide in marine habitats, but comprehensive physiological studies of chemoautotrophic bacteria thriving on animals are scarce. Stilbonematinae are coated by thiotrophic . As these nematodes migrate through the redox zone, their ectosymbionts experience varying oxygen concentrations. However, nothing is known about how these variations affect their physiology. Here, by applying omics, Raman microspectroscopy, and stable isotope labeling, we investigated the effect of oxygen on " Thiosymbion oneisti." Unexpectedly, sulfur oxidation genes were upregulated in anoxic relative to oxic conditions, but carbon fixation genes and incorporation of C-labeled bicarbonate were not. Instead, several genes involved in carbon fixation were upregulated under oxic conditions, together with genes involved in organic carbon assimilation, polyhydroxyalkanoate (PHA) biosynthesis, nitrogen fixation, and urea utilization. Furthermore, in the presence of oxygen, stress-related genes were upregulated together with vitamin biosynthesis genes likely necessary to withstand oxidative stress, and the symbiont appeared to proliferate less. Based on its physiological response to oxygen, we propose that " T. oneisti" may exploit anaerobic sulfur oxidation coupled to denitrification to proliferate in anoxic sand. However, the ectosymbiont would still profit from the oxygen available in superficial sand, as the energy-efficient aerobic respiration would facilitate carbon and nitrogen assimilation. Chemoautotrophic endosymbionts are famous for exploiting sulfur oxidization to feed marine organisms with fixed carbon. However, the physiology of thiotrophic bacteria thriving on the surface of animals (ectosymbionts) is less understood. One longstanding hypothesis posits that attachment to animals that migrate between reduced and oxic environments would boost sulfur oxidation, as the ectosymbionts would alternatively access sulfide and oxygen, the most favorable electron acceptor. Here, we investigated the effect of oxygen on the physiology of " Thiosymbion oneisti," a gammaproteobacterium which lives attached to marine nematodes inhabiting shallow-water sand. Surprisingly, sulfur oxidation genes were upregulated under anoxic relative to oxic conditions. Furthermore, under anoxia, the ectosymbiont appeared to be less stressed and to proliferate more. We propose that animal-mediated access to oxygen, rather than enhancing sulfur oxidation, would facilitate assimilation of carbon and nitrogen by the ectosymbiont.
Genomic insights into diverse bacterial taxa that degrade extracellular DNA in marine sediments
2021 - Nat Microbiol, 6: 885–898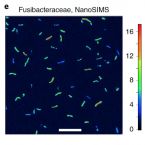
Abstract:
Extracellular DNA is a major macromolecule in global element cycles, and is a particularly crucial phosphorus, nitrogen and carbon source for microorganisms in the seafloor. Nevertheless, the identities, ecophysiology and genetic features of DNA-foraging microorganisms in marine sediments are largely unknown. Here, we combined microcosm experiments, DNA stable isotope probing (SIP), single-cell SIP using nano-scale secondary isotope mass spectrometry (NanoSIMS) and genome-centric metagenomics to study microbial catabolism of DNA and its subcomponents in marine sediments. 13C-DNA added to sediment microcosms was largely degraded within 10 d and mineralized to 13CO2. SIP probing of DNA revealed diverse ‘Candidatus Izemoplasma’, Lutibacter, Shewanella and Fusibacteraceae incorporated DNA-derived 13C-carbon. NanoSIMS confirmed incorporation of 13C into individual bacterial cells of Fusibacteraceae sorted from microcosms. Genomes of the 13C-labelled taxa all encoded enzymatic repertoires for catabolism of DNA or subcomponents of DNA. Comparative genomics indicated that diverse ‘Candidatus Izemoplasmatales’ (former Tenericutes) are exceptional because they encode multiple (up to five) predicted extracellular nucleases and are probably specialized DNA-degraders. Analyses of additional sediment metagenomes revealed extracellular nuclease genes are prevalent among Bacteroidota at diverse sites. Together, our results reveal the identities and functional properties of microorganisms that may contribute to the key ecosystem function of degrading and recycling DNA in the seabed.
Recently photoassimilated carbon and fungus-delivered nitrogen are spatially correlated in the ectomycorrhizal tissue of Fagus sylvatica.
2021 - New Phytol, 6: 2457-2474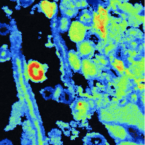
Abstract:
Ectomycorrhizal plants trade plant-assimilated carbon for soil nutrients with their fungal partners. The underlying mechanisms, however, are not fully understood. Here we investigate the exchange of carbon for nitrogen in the ectomycorrhizal symbiosis of Fagus sylvatica across different spatial scales from the root system to the cellular level. We provided N-labelled nitrogen to mycorrhizal hyphae associated with one half of the root system of young beech trees, while exposing plants to a CO atmosphere. We analysed the short-term distribution of C and N in the root system with isotope-ratio mass spectrometry, and at the cellular scale within a mycorrhizal root tip with nanoscale secondary ion mass spectrometry (NanoSIMS). At the root system scale, plants did not allocate more C to root parts that received more N. Nanoscale secondary ion mass spectrometry imaging, however, revealed a highly heterogenous, and spatially significantly correlated distribution of C and N at the cellular scale. Our results indicate that, on a coarse scale, plants do not allocate a larger proportion of photoassimilated C to root parts associated with N-delivering ectomycorrhizal fungi. Within the ectomycorrhizal tissue, however, recently plant-assimilated C and fungus-delivered N were spatially strongly coupled. Here, NanoSIMS visualisation provides an initial insight into the regulation of ectomycorrhizal C and N exchange at the microscale.
Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum.
2021 - Angew Chem Int Ed Engl, 60: 5063-5068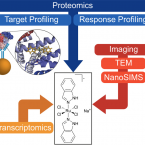
Abstract:
The ruthenium-based anticancer agent BOLD-100/KP1339 has shown promising results in several in vitro and in vivo tumour models as well as in early clinical trials. However, its mode of action remains to be fully elucidated. Recent evidence identified stress induction in the endoplasmic reticulum (ER) and concomitant down-modulation of HSPA5 (GRP78) as key drug effects. By exploiting the naturally formed adduct between BOLD-100 and human serum albumin as an immobilization strategy, we were able to perform target-profiling experiments that revealed the ribosomal proteins RPL10, RPL24, and the transcription factor GTF2I as potential interactors of this ruthenium(III) anticancer agent. Integrating these findings with proteomic profiling and transcriptomic experiments supported ribosomal disturbance and concomitant induction of ER stress. The formation of polyribosomes and ER swelling of treated cancer cells revealed by TEM validated this finding. Thus, the direct interaction of BOLD-100 with ribosomal proteins seems to accompany ER stress-induction and modulation of GRP78 in cancer cells.
Prevalence of RT-qPCR-detected SARS-CoV-2 infection at schools: First results from the Austrian School-SARS-CoV-2 prospective cohort study.
2021 - Lancet Reg Health Eur, 100086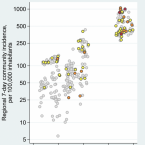
Abstract:
The role of schools in the SARS-CoV-2 pandemic is much debated. We aimed to quantify reliably the prevalence of SARS-CoV-2 infections at schools detected with reverse-transcription quantitative polymerase-chain-reaction (RT-qPCR).
This nationwide prospective cohort study monitors a representative sample of pupils (grade 1-8) and teachers at Austrian schools throughout the school year 2020/2021. We repeatedly test participants for SARS-CoV-2 infection using a gargling solution and RT-qPCR. We herein report on the first two rounds of examinations. We used mixed-effects logistic regression to estimate odds ratios and robust 95% confidence intervals (95% CI).
We analysed data on 10,734 participants from 245 schools (9465 pupils, 1269 teachers). Prevalence of SARS-CoV-2 infection increased from 0·39% at round 1 (95% CI 028-0·55%, 28 September-22 October 2020) to 1·39% at round 2 (95% CI 1·04-1·85%, 10-16 November). Odds ratios for SARS-CoV-2 infection were 2·26 (95% CI 1·25-4·12, = 0·007) in regions with >500 vs. ≤500 inhabitants/km, 1·67 (95% CI 1·42-1·97, <0·001) per two-fold higher regional 7-day community incidence, and 2·78 (95% CI 1·73-4·48, <0·001) in pupils at schools with high/very high vs. low/moderate social deprivation. Associations of regional community incidence and social deprivation persisted in a multivariable adjusted model. Prevalence did not differ by average number of pupils per class nor between age groups, sexes, pupils vs. teachers, or primary (grade 1-4) vs. secondary schools (grade 5-8).
This monitoring study in Austrian schools revealed SARS-CoV-2 infection in 0·39%-1·39% of participants and identified associations of regional community incidence and social deprivation with higher prevalence.
BMBWF Austria.Nano-scale imaging of dual stable isotope labeled oxaliplatin in human colon cancer cells reveals the nucleolus as a putative node for therapeutic effect
2021 - Nanoscale Advances, 3: 249-262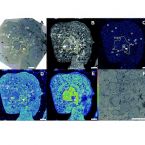
Abstract:
Oxaliplatin shows a superior clinical activity in colorectal cancer compared to cisplatin. Nevertheless, the knowledge about its cellular distribution and the mechanisms responsible for the different range of oxaliplatin-responsive tumors is far from complete. In this study, we combined highly sensitive element specific and isotope selective imaging by nanometer-scale secondary ion mass spectrometry (NanoSIMS) with transmission electron microscopy to investigate the subcellular accumulation of oxaliplatin in three human colon cancer cell lines (SW480, HCT116 wt, HCT116 OxR). Oxaliplatin bearing dual stable isotope labeled moieties, i.e. 2H-labeled diaminocyclohexane (DACH) and 13C-labeled oxalate, were applied for comparative analysis of the subcellular distribution patterns of the central metal and the ligands. In all the investigated cell lines, oxaliplatin was found to have a pronounced tendency for cytoplasmic aggregation in single membrane bound organelles, presumably related to various stages of the endocytic pathway. Moreover, nuclear structures, heterochromatin and in particular nucleoli, were affected by platinum-drug exposure. In order to explore the consequences of oxaliplatin resistance, subcellular drug distribution patterns were investigated in a pair of isogenic malignant cell lines with distinct levels of drug sensitivity (HCT116 wt and HCT116 OxR, the latter with acquired resistance to oxaliplatin). The subcellular platinum distribution was found to be similar in both cell lines, with only slightly higher accumulation in the sensitive HCT116 wt cells which is inconsistent with the resistance factor of more than 20-fold. Instead, the isotopic analysis revealed a disproportionally high accumulation of the oxalate ligand in the resistant cell line.
Optofluidic Raman-activated cell sorting for targeted genome retrieval or cultivation of microbial cells with specific functions.
2021 - Nat Protoc, 2: 634-676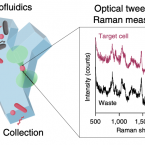
Abstract:
Stable isotope labeling of microbial taxa of interest and their sorting provide an efficient and direct way to answer the question "who does what?" in complex microbial communities when coupled with fluorescence in situ hybridization or downstream 'omics' analyses. We have developed a platform for automated Raman-based sorting in which optical tweezers and microfluidics are used to sort individual cells of interest from microbial communities on the basis of their Raman spectra. This sorting of cells and their downstream DNA analysis, such as by mini-metagenomics or single-cell genomics, or cultivation permits a direct link to be made between the metabolic roles and the genomes of microbial cells within complex microbial communities, as well as targeted isolation of novel microbes with a specific physiology of interest. We describe a protocol from sample preparation through Raman-activated live cell sorting. Subsequent cultivation of sorted cells is described, whereas downstream DNA analysis involves well-established approaches with abundant methods available in the literature. Compared with manual sorting, this technique provides a substantially higher throughput (up to 500 cells per h). Furthermore, the platform has very high sorting accuracy (98.3 ± 1.7%) and is fully automated, thus avoiding user biases that might accompany manual sorting. We anticipate that this protocol will empower in particular environmental and host-associated microbiome research with a versatile tool to elucidate the metabolic contributions of microbial taxa within their complex communities. After a 1-d preparation of cells, sorting takes on the order of 4 h, depending on the number of cells required.
Genomic and kinetic analysis of novel Nitrospinae enriched by cell sorting.
2021 - ISME J, 15: 732–745
Abstract:
Chemolithoautotrophic nitrite-oxidizing bacteria (NOB) are key players in global nitrogen and carbon cycling. Members of the phylum Nitrospinae are the most abundant, known NOB in the oceans. To date, only two closely affiliated Nitrospinae species have been isolated, which are only distantly related to the environmentally abundant uncultured Nitrospinae clades. Here, we applied live cell sorting, activity screening, and subcultivation on marine nitrite-oxidizing enrichments to obtain novel marine Nitrospinae. Two binary cultures were obtained, each containing one Nitrospinae strain and one alphaproteobacterial heterotroph. The Nitrospinae strains represent two new genera, and one strain is more closely related to environmentally abundant Nitrospinae than previously cultured NOB. With an apparent half-saturation constant of 8.7 ± 2.5 µM, this strain has the highest affinity for nitrite among characterized marine NOB, while the other strain (16.2 ± 1.6 µM) and Nitrospina gracilis (20.1 ± 2.1 µM) displayed slightly lower nitrite affinities. The new strains and N. gracilis share core metabolic pathways for nitrite oxidation and CO fixation but differ remarkably in their genomic repertoires of terminal oxidases, use of organic N sources, alternative energy metabolisms, osmotic stress and phage defense. The new strains, tentatively named "Candidatus Nitrohelix vancouverensis" and "Candidatus Nitronauta litoralis", shed light on the niche differentiation and potential ecological roles of Nitrospinae.
Flow-through stable isotope probing (Flow-SIP) minimizes cross-feeding in complex microbial communities.
2021 - ISME J, 1: 348-353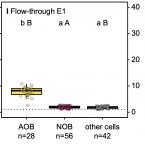
Abstract:
Stable isotope probing (SIP) is a key tool for identifying the microorganisms catalyzing the turnover of specific substrates in the environment and to quantify their relative contributions to biogeochemical processes. However, SIP-based studies are subject to the uncertainties posed by cross-feeding, where microorganisms release isotopically labeled products, which are then used by other microorganisms, instead of incorporating the added tracer directly. Here, we introduce a SIP approach that has the potential to strongly reduce cross-feeding in complex microbial communities. In this approach, the microbial cells are exposed on a membrane filter to a continuous flow of medium containing isotopically labeled substrate. Thereby, metabolites and degradation products are constantly removed, preventing consumption of these secondary substrates. A nanoSIMS-based proof-of-concept experiment using nitrifiers in activated sludge and C-bicarbonate as an activity tracer showed that Flow-SIP significantly reduces cross-feeding and thus allows distinguishing primary consumers from other members of microbial food webs.
Rational design of a microbial consortium of mucosal sugar utilizers reduces Clostridiodes difficile colonization.
2020 - Nat Commun, 1: 5104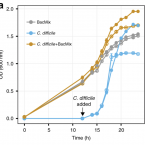
Abstract:
Many intestinal pathogens, including Clostridioides difficile, use mucus-derived sugars as crucial nutrients in the gut. Commensals that compete with pathogens for such nutrients are therefore ecological gatekeepers in healthy guts, and are attractive candidates for therapeutic interventions. Nevertheless, there is a poor understanding of which commensals use mucin-derived sugars in situ as well as their potential to impede pathogen colonization. Here, we identify mouse gut commensals that utilize mucus-derived monosaccharides within complex communities using single-cell stable isotope probing, Raman-activated cell sorting and mini-metagenomics. Sequencing of cell-sorted fractions reveals members of the underexplored family Muribaculaceae as major mucin monosaccharide foragers, followed by members of Lachnospiraceae, Rikenellaceae, and Bacteroidaceae families. Using this information, we assembled a five-member consortium of sialic acid and N-acetylglucosamine utilizers that impedes C. difficile's access to these mucosal sugars and impairs pathogen colonization in antibiotic-treated mice. Our findings underscore the value of targeted approaches to identify organisms utilizing key nutrients and to rationally design effective probiotic mixtures.
Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities
2020 - Int J Syst Evol Microbiol, 11: 5972-6016
Abstract:
The class comprises an ecologically and metabolically diverse group of bacteria best known for dissimilatory sulphate reduction and predatory behaviour. Although this lineage is the fourth described class of the phylum , it rarely affiliates with other proteobacterial classes and is frequently not recovered as a monophyletic unit in phylogenetic analyses. Indeed, one branch of the class encompassing like predators was recently reclassified into a separate proteobacterial class, the . Here we systematically explore the phylogeny of taxa currently assigned to these classes using 120 conserved single-copy marker genes as well as rRNA genes. The overwhelming majority of markers reject the inclusion of the classes and in the phylum . Instead, the great majority of currently recognized members of the class are better classified into four novel phylum-level lineages. We propose the names phyl. nov. and phyl. nov. for two of these phyla, based on the oldest validly published names in each lineage, and retain the placeholder name SAR324 for the third phylum pending formal description of type material. Members of the class represent a separate phylum for which we propose the name phyl. nov. based on priority in the literature and general recognition of the genus phyl. nov. includes the taxa previously classified in the phylum , and these reclassifications imply that the ability of sulphate reduction was vertically inherited in the rather than laterally acquired as previously inferred. Our analysis also indicates the independent acquisition of predatory behaviour in the phyla and , which is consistent with their distinct modes of action. This work represents a stable reclassification of one of the most taxonomically challenging areas of the bacterial tree and provides a robust framework for future ecological and systematic studies.
A refined set of rRNA-targeted oligonucleotide probes for in situ detection and quantification of ammonia-oxidizing bacteria
2020 - Water Res., 186: 116372
Abstract:
Ammonia-oxidizing bacteria (AOB) of the betaproteobacterial genera Nitrosomonas and Nitrosospira are key nitrifying microorganisms in many natural and engineered ecosystems. Since many AOB remain uncultured, fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes has been one of the most widely used approaches to study the community composition, abundance, and other features of AOB directly in environmental samples. However, the established and widely used AOB-specific 16S rRNA-targeted FISH probes were designed up to two decades ago, based on much smaller rRNA gene sequence datasets than available today. Several of these probes cover their target AOB lineages incompletely and suffer from a weak target specificity, which causes cross-hybridization of probes that should detect different AOB lineages. Here, a set of new highly specific 16S rRNA-targeted oligonucleotide probes was developed and experimentally evaluated that complements the existing probes and enables the specific detection and differentiation of the known, major phylogenetic clusters of betaproteobacterial AOB. The new probes were successfully applied to visualize and quantify AOB in activated sludge and biofilm samples from seven pilot- and full-scale wastewater treatment systems. Based on its improved target group coverage and specificity, the refined probe set will facilitate future in situ analyses of AOB.
Exploring the upper pH limits of nitrite oxidation: diversity, ecophysiology, and adaptive traits of haloalkalitolerant Nitrospira.
2020 - ISME J, 12: 2967-2979
Abstract:
Nitrite-oxidizing bacteria of the genus Nitrospira are key players of the biogeochemical nitrogen cycle. However, little is known about their occurrence and survival strategies in extreme pH environments. Here, we report on the discovery of physiologically versatile, haloalkalitolerant Nitrospira that drive nitrite oxidation at exceptionally high pH. Nitrospira distribution, diversity, and ecophysiology were studied in hypo- and subsaline (1.3-12.8 g salt/l), highly alkaline (pH 8.9-10.3) lakes by amplicon sequencing, metagenomics, and cultivation-based approaches. Surprisingly, not only were Nitrospira populations detected, but they were also considerably diverse with presence of members from Nitrospira lineages I, II and IV. Furthermore, the ability of Nitrospira enrichment cultures to oxidize nitrite at neutral to highly alkaline pH of 10.5 was demonstrated. Metagenomic analysis of a newly enriched Nitrospira lineage IV species, "Candidatus Nitrospira alkalitolerans", revealed numerous adaptive features of this organism to its extreme environment. Among them were a sodium-dependent N-type ATPase and NADH:quinone oxidoreductase next to the proton-driven forms usually found in Nitrospira. Other functions aid in pH and cation homeostasis and osmotic stress defense. "Ca. Nitrospira alkalitolerans" also possesses group 2a and 3b [NiFe] hydrogenases, suggesting it can use hydrogen as alternative energy source. These results reveal how Nitrospira cope with strongly fluctuating pH and salinity conditions and expand our knowledge of nitrogen cycling in extreme habitats.
Composition and activity of nitrifier communities in soil are unresponsive to elevated temperature and CO, but strongly affected by drought.
2020 - ISME J, 12: 3038-3053
Abstract:
Nitrification is a fundamental process in terrestrial nitrogen cycling. However, detailed information on how climate change affects the structure of nitrifier communities is lacking, specifically from experiments in which multiple climate change factors are manipulated simultaneously. Consequently, our ability to predict how soil nitrogen (N) cycling will change in a future climate is limited. We conducted a field experiment in a managed grassland and simultaneously tested the effects of elevated atmospheric CO, temperature, and drought on the abundance of active ammonia-oxidizing bacteria (AOB) and archaea (AOA), comammox (CMX) Nitrospira, and nitrite-oxidizing bacteria (NOB), and on gross mineralization and nitrification rates. We found that N transformation processes, as well as gene and transcript abundances, and nitrifier community composition were remarkably resistant to individual and interactive effects of elevated CO and temperature. During drought however, process rates were increased or at least maintained. At the same time, the abundance of active AOB increased probably due to higher NH availability. Both, AOA and comammox Nitrospira decreased in response to drought and the active community composition of AOA and NOB was also significantly affected. In summary, our findings suggest that warming and elevated CO have only minor effects on nitrifier communities and soil biogeochemical variables in managed grasslands, whereas drought favors AOB and increases nitrification rates. This highlights the overriding importance of drought as a global change driver impacting on soil microbial community structure and its consequences for N cycling.
Roadmap for naming uncultivated Archaea and Bacteria.
2020 - Nat Microbiol, 8: 987-994
Abstract:
The assembly of single-amplified genomes (SAGs) and metagenome-assembled genomes (MAGs) has led to a surge in genome-based discoveries of members affiliated with Archaea and Bacteria, bringing with it a need to develop guidelines for nomenclature of uncultivated microorganisms. The International Code of Nomenclature of Prokaryotes (ICNP) only recognizes cultures as 'type material', thereby preventing the naming of uncultivated organisms. In this Consensus Statement, we propose two potential paths to solve this nomenclatural conundrum. One option is the adoption of previously proposed modifications to the ICNP to recognize DNA sequences as acceptable type material; the other option creates a nomenclatural code for uncultivated Archaea and Bacteria that could eventually be merged with the ICNP in the future. Regardless of the path taken, we believe that action is needed now within the scientific community to develop consistent rules for nomenclature of uncultivated taxa in order to provide clarity and stability, and to effectively communicate microbial diversity.
Raman-based sorting of microbial cells to link functions to their genes.
2020 - Microb Cell, 3: 62-65
Abstract:
In our recent work, we developed an optofluidic platform that allows a direct link to be made between the phenotypes (functions) and the genotypes (genes) of microbial cells within natural communities. By combining stable isotope probing, optical tweezers, Raman microspectroscopy, and microfluidics, the platform performs automated Raman-based sorting of taxa from within a complex community in terms of their functional properties. In comparison with manual sorting approaches, our method provides high throughput (up to 500 cells per hour) and very high sorting accuracy (98.3 ± 1.7%), and significantly reduces the human labour required. The system provides an efficient manner to untangle the contributions of individual members within environmental and host-associated microbiomes. In this News and Thoughts, we provide an overview of our platform, describe potential applications, suggest ways in which the system could be improved, and discuss future directions in which Raman-based analysis of microbial populations might be developed.
Microbiome definition re-visited: old concepts and new challenges.
2020 - Microbiome, 1: 103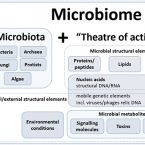
Abstract:
The field of microbiome research has evolved rapidly over the past few decades and has become a topic of great scientific and public interest. As a result of this rapid growth in interest covering different fields, we are lacking a clear commonly agreed definition of the term "microbiome." Moreover, a consensus on best practices in microbiome research is missing. Recently, a panel of international experts discussed the current gaps in the frame of the European-funded MicrobiomeSupport project. The meeting brought together about 40 leaders from diverse microbiome areas, while more than a hundred experts from all over the world took part in an online survey accompanying the workshop. This article excerpts the outcomes of the workshop and the corresponding online survey embedded in a short historical introduction and future outlook. We propose a definition of microbiome based on the compact, clear, and comprehensive description of the term provided by Whipps et al. in 1988, amended with a set of novel recommendations considering the latest technological developments and research findings. We clearly separate the terms microbiome and microbiota and provide a comprehensive discussion considering the composition of microbiota, the heterogeneity and dynamics of microbiomes in time and space, the stability and resilience of microbial networks, the definition of core microbiomes, and functionally relevant keystone species as well as co-evolutionary principles of microbe-host and inter-species interactions within the microbiome. These broad definitions together with the suggested unifying concepts will help to improve standardization of microbiome studies in the future, and could be the starting point for an integrated assessment of data resulting in a more rapid transfer of knowledge from basic science into practice. Furthermore, microbiome standards are important for solving new challenges associated with anthropogenic-driven changes in the field of planetary health, for which the understanding of microbiomes might play a key role. Video Abstract.
Single cell analyses reveal contrasting life strategies of the two main nitrifiers in the ocean.
2020 - Nat Commun, 1: 767
Abstract:
Nitrification, the oxidation of ammonia via nitrite to nitrate, is a key process in marine nitrogen (N) cycling. Although oceanic ammonia and nitrite oxidation are balanced, ammonia-oxidizing archaea (AOA) vastly outnumber the main nitrite oxidizers, the bacterial Nitrospinae. The ecophysiological reasons for this discrepancy in abundance are unclear. Here, we compare substrate utilization and growth of Nitrospinae to AOA in the Gulf of Mexico. Based on our results, more than half of the Nitrospinae cellular N-demand is met by the organic-N compounds urea and cyanate, while AOA mainly assimilate ammonium. Nitrospinae have, under in situ conditions, around four-times higher biomass yield and five-times higher growth rates than AOA, despite their ten-fold lower abundance. Our combined results indicate that differences in mortality between Nitrospinae and AOA, rather than thermodynamics, biomass yield and cell size, determine the abundances of these main marine nitrifiers. Furthermore, there is no need to invoke yet undiscovered, abundant nitrite oxidizers to explain nitrification rates in the ocean.
Transcriptomic Response of Nitrosomonas europaea Transitioned from Ammonia- to Oxygen-Limited Steady-State Growth.
2020 - mSystems, 1: e00562-19
Abstract:
Ammonia-oxidizing microorganisms perform the first step of nitrification, the oxidation of ammonia to nitrite. The bacterium is the best-characterized ammonia oxidizer to date. Exposure to hypoxic conditions has a profound effect on the physiology of , e.g., by inducing nitrifier denitrification, resulting in increased nitric and nitrous oxide production. This metabolic shift is of major significance in agricultural soils, as it contributes to fertilizer loss and global climate change. Previous studies investigating the effect of oxygen limitation on have focused on the transcriptional regulation of genes involved in nitrification and nitrifier denitrification. Here, we combine steady-state cultivation with whole-genome transcriptomics to investigate the overall effect of oxygen limitation on Under oxygen-limited conditions, growth yield was reduced and ammonia-to-nitrite conversion was not stoichiometric, suggesting the production of nitrogenous gases. However, the transcription of the principal nitric oxide reductase (cNOR) did not change significantly during oxygen-limited growth, while the transcription of the nitrite reductase-encoding gene () was significantly lower. In contrast, both heme-copper-containing cytochrome oxidases encoded by were upregulated during oxygen-limited growth. Particularly striking was the significant increase in transcription of the B-type heme-copper oxidase, proposed to function as a nitric oxide reductase (sNOR) in ammonia-oxidizing bacteria. In the context of previous physiological studies, as well as the evolutionary placement of sNOR with regard to other heme-copper oxidases, these results suggest sNOR may function as a high-affinity terminal oxidase in and other ammonia-oxidizing bacteria. Nitrification is a ubiquitous microbially mediated process in the environment and an essential process in engineered systems such as wastewater and drinking water treatment plants. However, nitrification also contributes to fertilizer loss from agricultural environments, increasing the eutrophication of downstream aquatic ecosystems, and produces the greenhouse gas nitrous oxide. As ammonia-oxidizing bacteria are the most dominant ammonia-oxidizing microbes in fertilized agricultural soils, understanding their responses to a variety of environmental conditions is essential for curbing the negative environmental effects of nitrification. Notably, oxygen limitation has been reported to significantly increase nitric oxide and nitrous oxide production during nitrification. Here, we investigate the physiology of the best-characterized ammonia-oxidizing bacterium, , growing under oxygen-limited conditions.
Archaeal nitrification is constrained by copper complexation with organic matter in municipal wastewater treatment plants.
2020 - ISME J, 2: 335-346
Abstract:
Consistent with the observation that ammonia-oxidizing bacteria (AOB) outnumber ammonia-oxidizing archaea (AOA) in many eutrophic ecosystems globally, AOB typically dominate activated sludge aeration basins from municipal wastewater treatment plants (WWTPs). In this study, we demonstrate that the growth of AOA strains inoculated into sterile-filtered wastewater was inhibited significantly, in contrast to uninhibited growth of a reference AOB strain. In order to identify possible mechanisms underlying AOA-specific inhibition, we show that complex mixtures of organic compounds, such as yeast extract, were highly inhibitory to all AOA strains but not to the AOB strain. By testing individual organic compounds, we reveal strong inhibitory effects of organic compounds with high metal complexation potentials implying that the inhibitory mechanism for AOA can be explained by the reduced bioavailability of an essential metal. Our results further demonstrate that the inhibitory effect on AOA can be alleviated by copper supplementation, which we observed for pure AOA cultures in a defined medium and for AOA inoculated into nitrifying sludge. Our study offers a novel mechanistic explanation for the relatively low abundance of AOA in most WWTPs and provides a basis for modulating the composition of nitrifying communities in both engineered systems and naturally occurring environments.
On the evolution and physiology of cable bacteria.
2019 - Proc. Natl. Acad. Sci. U.S.A., 38: 19116-19125
Abstract:
Cable bacteria of the family Desulfobulbaceae form centimeter-long filaments comprising thousands of cells. They occur worldwide in the surface of aquatic sediments, where they connect sulfide oxidation with oxygen or nitrate reduction via long-distance electron transport. In the absence of pure cultures, we used single-filament genomics and metagenomics to retrieve draft genomes of 3 marine Electrothrix and 1 freshwater Electronema species. These genomes contain >50% unknown genes but still share their core genomic makeup with sulfate-reducing and sulfur-disproportionating Desulfobulbaceae, with few core genes lost and 212 unique genes (from 197 gene families) conserved among cable bacteria. Last common ancestor analysis indicates gene divergence and lateral gene transfer as equally important origins of these unique genes. With support from metaproteomics of a Electronema enrichment, the genomes suggest that cable bacteria oxidize sulfide by reversing the canonical sulfate reduction pathway and fix CO using the Wood-Ljungdahl pathway. Cable bacteria show limited organotrophic potential, may assimilate smaller organic acids and alcohols, fix N, and synthesize polyphosphates and polyglucose as storage compounds; several of these traits were confirmed by cell-level experimental analyses. We propose a model for electron flow from sulfide to oxygen that involves periplasmic cytochromes, yet-unidentified conductive periplasmic fibers, and periplasmic oxygen reduction. This model proposes that an active cable bacterium gains energy in the anodic, sulfide-oxidizing cells, whereas cells in the oxic zone flare off electrons through intense cathodic oxygen respiration without energy conservation; this peculiar form of multicellularity seems unparalleled in the microbial world.
Membrane Lipid Composition of the Moderately Thermophilic Ammonia-Oxidizing Archaeon " Nitrosotenuis uzonensis" at Different Growth Temperatures.
2019 - Appl. Environ. Microbiol., 20: 1–17
Abstract:
" Nitrosotenuis uzonensis" is the only cultured moderately thermophilic member of the thaumarchaeotal order (NP) that contains many mesophilic marine strains. We examined its membrane lipid composition at different growth temperatures (37°C, 46°C, and 50°C). Its lipids were all membrane-spanning glycerol dialkyl glycerol tetraethers (GDGTs), with 0 to 4 cyclopentane moieties. Crenarchaeol (cren), the characteristic thaumarchaeotal GDGT, and its isomer (cren') were present in high abundance (30 to 70%). The GDGT polar headgroups were mono-, di-, and trihexoses and hexose/phosphohexose. The ratio of glycolipid to phospholipid GDGTs was highest in the cultures grown at 50°C. With increasing growth temperatures, the relative contributions of cren and cren' increased, while those of GDGT-0 to GDGT-4 (including isomers) decreased. TEX (tetraether index of tetraethers consisting of 86 carbons)-derived temperatures were much lower than the actual growth temperatures, further demonstrating that TEX does not accurately reflect the membrane lipid adaptation of thermophilic As the temperature increased, specific GDGTs changed relative to their isomers, possibly representing temperature adaption-induced changes in cyclopentane ring stereochemistry. Comparison of a wide range of thaumarchaeotal core lipid compositions revealed that the " Nitrosotenuis uzonensis" cultures clustered separately from other members of the NP order and the (NS) order. While phylogeny generally seems to have a strong influence on GDGT distribution, our analysis of " Nitrosotenuis uzonensis" demonstrates that its terrestrial, higher-temperature niche has led to a lipid composition that clearly differentiates it from other NP members and that this difference is mostly driven by its high cren' content. For , the ratio of their glycerol dialkyl glycerol tetraether (GDGT) lipids depends on growth temperature, a premise that forms the basis of the widely applied TEX paleotemperature proxy. A thorough understanding of which GDGTs are produced by which and what the effect of temperature is on their GDGT composition is essential for constraining the TEX proxy. " Nitrosotenuis uzonensis" is a moderately thermophilic thaumarchaeote enriched from a thermal spring, setting it apart in its environmental niche from the other marine mesophilic members of its order. Indeed, we found that the GDGT composition of " Nitrosotenuis uzonensis" cultures was distinct from those of other members of its order and was more similar to those of other thermophilic, terrestrial This suggests that while phylogeny has a strong influence on GDGT distribution, the environmental niche that a thaumarchaeote inhabits also shapes its GDGT composition.
Expansion of Thaumarchaeota habitat range is correlated with horizontal transfer of ATPase operons.
2019 - ISME J, 12: 3067-3079
Abstract:
Thaumarchaeota are responsible for a significant fraction of ammonia oxidation in the oceans and in soils that range from alkaline to acidic. However, the adaptive mechanisms underpinning their habitat expansion remain poorly understood. Here we show that expansion into acidic soils and the high pressures of the hadopelagic zone of the oceans is tightly linked to the acquisition of a variant of the energy-yielding ATPases via horizontal transfer. Whereas the ATPase genealogy of neutrophilic Thaumarchaeota is congruent with their organismal genealogy inferred from concatenated conserved proteins, a common clade of V-type ATPases unites phylogenetically distinct clades of acidophilic/acid-tolerant and piezophilic/piezotolerant species. A presumptive function of pumping cytoplasmic protons at low pH is consistent with the experimentally observed increased expression of the V-ATPase in an acid-tolerant thaumarchaeote at low pH. Consistently, heterologous expression of the thaumarchaeotal V-ATPase significantly increased the growth rate of E. coli at low pH. Its adaptive significance to growth in ocean trenches may relate to pressure-related changes in membrane structure in which this complex molecular machine must function. Together, our findings reveal that the habitat expansion of Thaumarchaeota is tightly correlated with extensive horizontal transfer of atp operons.
Specific micropollutant biotransformation pattern by the comammox bacterium Nitrospira inopinata
2019 - Environ. Sci. Technol., 15: 8695-8705
Abstract:
The recently discovered complete ammonia-oxidizing (comammox) bacteria occur in various environments, including wastewater treatment plants. To better understand their role in micropollutant biotransformation in comparison with ammonia-oxidizing bacteria (AOB) and ammonia-oxidizing archaea (AOA), we investigated the biotransformation capability of (the only comammox isolate) for 17 micropollutants. Asulam, fenhexamid, mianserin, and ranitidine were biotransformed by , (AOA), and Nm90 (AOB). More distinctively, carbendazim, a benzimidazole fungicide, was exclusively biotransformed by . The biotransformation of carbendazim only occurred when was supplied with ammonia but not nitrite as the energy source. The exclusive biotransformation of carbendazim by was likely enabled by an enhanced substrate promiscuity of its unique AMO and its much higher substrate (for ammonia) affinity compared with the other two ammonia oxidizers. One major plausible transformation product (TP) of carbendazim is a hydroxylated form at the aromatic ring, which is consistent with the function of AMO. These findings provide fundamental knowledge on the micropollutant degradation potential of a comammox bacterium to better understand the fate of micropollutants in nitrifying environments.
Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella basta.
2019 - Environ. Microbiol., 10: 3831-3854
Abstract:
Marine sponges represent one of the few eukaryotic groups that frequently harbour symbiotic members of the Thaumarchaeota, which are important chemoautotrophic ammonia-oxidizers in many environments. However, in most studies, direct demonstration of ammonia-oxidation by these archaea within sponges is lacking, and little is known about sponge-specific adaptations of ammonia-oxidizing archaea (AOA). Here, we characterized the thaumarchaeal symbiont of the marine sponge Ianthella basta using metaproteogenomics, fluorescence in situ hybridization, qPCR and isotope-based functional assays. 'Candidatus Nitrosospongia ianthellae' is only distantly related to cultured AOA. It is an abundant symbiont that is solely responsible for nitrite formation from ammonia in I. basta that surprisingly does not harbour nitrite-oxidizing microbes. Furthermore, this AOA is equipped with an expanded set of extracellular subtilisin-like proteases, a metalloprotease unique among archaea, as well as a putative branched-chain amino acid ABC transporter. This repertoire is strongly indicative of a mixotrophic lifestyle and is (with slight variations) also found in other sponge-associated, but not in free-living AOA. We predict that this feature as well as an expanded and unique set of secreted serpins (protease inhibitors), a unique array of eukaryotic-like proteins, and a DNA-phosporothioation system, represent important adaptations of AOA to life within these ancient filter-feeding animals.
Indications for enzymatic denitrification to N2O at low pH in an ammonia-oxidizing archaeon.
2019 - ISME J, 13: 2633-2638
Abstract:
Nitrous oxide (NO) is a key climate change gas and nitrifying microbes living in terrestrial ecosystems contribute significantly to its formation. Many soils are acidic and global change will cause acidification of aquatic and terrestrial ecosystems, but the effect of decreasing pH on NO formation by nitrifiers is poorly understood. Here, we used isotope-ratio mass spectrometry to investigate the effect of acidification on production of NO by pure cultures of two ammonia-oxidizing archaea (AOA; Nitrosocosmicus oleophilus and Nitrosotenuis chungbukensis) and an ammonia-oxidizing bacterium (AOB; Nitrosomonas europaea). For all three strains acidification led to increased emission of NO. However, changes of N site preference (SP) values within the NO molecule (as indicators of pathways for NO formation), caused by decreasing pH, were highly different between the tested AOA and AOB. While acidification decreased the SP value in the AOB strain, SP values increased to a maximum value of 29‰ in N. oleophilus. In addition, N-nitrite tracer experiments showed that acidification boosted nitrite transformation into NO in all strains, but the incorporation rate was different for each ammonia oxidizer. Unexpectedly, for N. oleophilus more than 50% of the NO produced at pH 5.5 had both nitrogen atoms from nitrite and we demonstrated that under these conditions expression of a putative cytochrome P450 NO reductase is strongly upregulated. Collectively, our results indicate that N. oleophilus might be able to enzymatically denitrify nitrite to NO at low pH.
Global diversity and biogeography of bacterial communities in wastewater treatment plants.
2019 - Nat Microbiol, 7: 1183-1195
Abstract:
Microorganisms in wastewater treatment plants (WWTPs) are essential for water purification to protect public and environmental health. However, the diversity of microorganisms and the factors that control it are poorly understood. Using a systematic global-sampling effort, we analysed the 16S ribosomal RNA gene sequences from ~1,200 activated sludge samples taken from 269 WWTPs in 23 countries on 6 continents. Our analyses revealed that the global activated sludge bacterial communities contain ~1 billion bacterial phylotypes with a Poisson lognormal diversity distribution. Despite this high diversity, activated sludge has a small, global core bacterial community (n = 28 operational taxonomic units) that is strongly linked to activated sludge performance. Meta-analyses with global datasets associate the activated sludge microbiomes most closely to freshwater populations. In contrast to macroorganism diversity, activated sludge bacterial communities show no latitudinal gradient. Furthermore, their spatial turnover is scale-dependent and appears to be largely driven by stochastic processes (dispersal and drift), although deterministic factors (temperature and organic input) are also important. Our findings enhance our mechanistic understanding of the global diversity and biogeography of activated sludge bacterial communities within a theoretical ecology framework and have important implications for microbial ecology and wastewater treatment processes.
Cometabolic biotransformation and microbial-mediated abiotic transformation of sulfonamides by three ammonia oxidizers.
2019 - Water Res., 444-453
Abstract:
The abilities of three phylogenetically distant ammonia oxidizers, Nitrososphaera gargensis, an ammonia-oxidizing archaeon (AOA); Nitrosomomas nitrosa Nm90, an ammonia-oxidizing bacterium (AOB); and Nitrospira inopinata, the only complete ammonia oxidizer (comammox) available as a pure culture, to biotransform seven sulfonamides (SAs) were investigated. The removals and protein-normalized biotransformation rate constants indicated that the AOA strain N. gargensis exhibited the highest SA biotransformation rates, followed by N. inopinata and N. nitrosa Nm90. The transformation products (TPs) of sulfadiazine (SDZ), sulfamethazine (SMZ) and sulfamethoxazole (SMX) and the biotransformation mechanisms were evaluated. Based on the analysis of the TP formulas and approximate structures, it was found that during biotransformation, i) the AOA strain carried out SA deamination, hydroxylation, and nitration; ii) the AOB strain mainly performed SA deamination; and iii) the comammox isolate participated only in deamination reactions. It is proposed that deamination was catalyzed by deaminases while hydroxylation and nitration were mediated by nonspecific activities of the ammonia monooxygenase (AMO). Additionally, it was demonstrated that among the three ammonia oxidizers, only AOB contributed to the formation of pterin-SA conjugates. The biotransformation of SDZ, SMZ and SMX occurred only when ammonia oxidation was active, suggesting a cometabolic transformation mechanism. Interestingly, SAs could also be transformed by hydroxylamine, an intermediate of ammonia oxidation, suggesting that in addition to enzymatic conversions, a microbially induced abiotic mechanism contributes to SA transformation during ammonia oxidation. Overall, using experiments with pure cultures, this study provides important insights into the roles played by ammonia oxidizers in SA biotransformation.
Low yield and abiotic origin of N2O formed by the complete nitrifier Nitrospira inopinata.
2019 - Nat Commun, 1: 1836
Abstract:
Nitrous oxide (NO) and nitric oxide (NO) are atmospheric trace gases that contribute to climate change and affect stratospheric and ground-level ozone concentrations. Ammonia oxidizing bacteria (AOB) and archaea (AOA) are key players in the nitrogen cycle and major producers of NO and NO globally. However, nothing is known about NO and NO production by the recently discovered and widely distributed complete ammonia oxidizers (comammox). Here, we show that the comammox bacterium Nitrospira inopinata is sensitive to inhibition by an NO scavenger, cannot denitrify to NO, and emits NO at levels that are comparable to AOA but much lower than AOB. Furthermore, we demonstrate that NO formed by N. inopinata formed under varying oxygen regimes originates from abiotic conversion of hydroxylamine. Our findings indicate that comammox microbes may produce less NO during nitrification than AOB.
Widespread soil bacterium that oxidizes atmospheric methane.
2019 - Proc. Natl. Acad. Sci. U.S.A., 17: 8515-8524
Abstract:
The global atmospheric level of methane (CH), the second most important greenhouse gas, is currently increasing by ∼10 million tons per year. Microbial oxidation in unsaturated soils is the only known biological process that removes CH from the atmosphere, but so far, bacteria that can grow on atmospheric CH have eluded all cultivation efforts. In this study, we have isolated a pure culture of a bacterium, strain MG08 that grows on air at atmospheric concentrations of CH [1.86 parts per million volume (p.p.m.v.)]. This organism, named , is globally distributed in soils and closely related to uncultured members of the upland soil cluster α. CH oxidation experiments and C-single cell isotope analyses demonstrated that it oxidizes atmospheric CH aerobically and assimilates carbon from both CH and CO Its estimated specific affinity for CH (a) is the highest for any cultivated methanotroph. However, growth on ambient air was also confirmed for and , close relatives with a lower specific affinity for CH, suggesting that the ability to utilize atmospheric CH for growth is more widespread than previously believed. The closed genome of MG08 encodes a single particulate methane monooxygenase, the serine cycle for assimilation of carbon from CH and CO, and CO fixation via the recently postulated reductive glycine pathway. It also fixes dinitrogen and expresses the genes for a high-affinity hydrogenase and carbon monoxide dehydrogenase, suggesting that atmospheric CH oxidizers harvest additional energy from oxidation of the atmospheric trace gases carbon monoxide (0.2 p.p.m.v.) and hydrogen (0.5 p.p.m.v.).
An automated Raman-based platform for the sorting of live cells by functional properties.
2019 - Nat Microbiol, 6: 1035-1048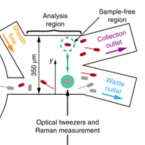
Abstract:
Stable-isotope probing is widely used to study the function of microbial taxa in their natural environment, but sorting of isotopically labelled microbial cells from complex samples for subsequent genomic analysis or cultivation is still in its early infancy. Here, we introduce an optofluidic platform for automated sorting of stable-isotope-probing-labelled microbial cells, combining microfluidics, optical tweezing and Raman microspectroscopy, which yields live cells suitable for subsequent single-cell genomics, mini-metagenomics or cultivation. We describe the design and optimization of this Raman-activated cell-sorting approach, illustrate its operation with four model bacteria (two intestinal, one soil and one marine) and demonstrate its high sorting accuracy (98.3 ± 1.7%), throughput (200-500 cells h; 3.3-8.3 cells min) and compatibility with cultivation. Application of this sorting approach for the metagenomic characterization of bacteria involved in mucin degradation in the mouse colon revealed a diverse consortium of bacteria, including several members of the underexplored family Muribaculaceae, highlighting both the complexity of this niche and the potential of Raman-activated cell sorting for identifying key players in targeted processes.
Resolving the individual contribution of key microbial populations to enhanced biological phosphorus removal with Raman-FISH.
2019 - ISME J, 8: 1933-1946
Abstract:
Enhanced biological phosphorus removal (EBPR) is a globally important biotechnological process and relies on the massive accumulation of phosphate within special microorganisms. Candidatus Accumulibacter conform to the classical physiology model for polyphosphate accumulating organisms and are widely believed to be the most important player for the process in full-scale EBPR systems. However, it was impossible till now to quantify the contribution of specific microbial clades to EBPR. In this study, we have developed a new tool to directly link the identity of microbial cells to the absolute quantification of intracellular poly-P and other polymers under in situ conditions, and applied it to eight full-scale EBPR plants. Besides Ca. Accumulibacter, members of the genus Tetrasphaera were found to be important microbes for P accumulation, and in six plants they were the most important. As these Tetrasphaera cells did not exhibit the classical phenotype of poly-P accumulating microbes, our entire understanding of the microbiology of the EBPR process has to be revised. Furthermore, our new single-cell approach can now also be applied to quantify storage polymer dynamics in individual populations in situ in other ecosystems and might become a valuable tool for many environmental microbiologists.
Rapid transfer of plant photosynthates to soil bacteria via ectomycorrhizal hyphae and its interaction with nitrogen availability.
2019 - Front Microbiol, 168
Abstract:
Plant roots release recent photosynthates into the rhizosphere, accelerating decomposition of organic matter by saprotrophic soil microbes ("rhizosphere priming effect") which consequently increases nutrient availability for plants. However, about 90% of all higher plant species are mycorrhizal, transferring a significant fraction of their photosynthates directly to their fungal partners. Whether mycorrhizal fungi pass on plant-derived carbon (C) to bacteria in root-distant soil areas, i.e., incite a "hyphosphere priming effect," is not known. Experimental evidence for C transfer from mycorrhizal hyphae to soil bacteria is limited, especially for ectomycorrhizal systems. As ectomycorrhizal fungi possess enzymatic capabilities to degrade organic matter themselves, it remains unclear whether they cooperate with soil bacteria by providing photosynthates, or compete for available nutrients. To investigate a possible C transfer from ectomycorrhizal hyphae to soil bacteria, and its response to changing nutrient availability, we planted young beech trees () into "split-root" boxes, dividing their root systems into two disconnected soil compartments. Each of these compartments was separated from a litter compartment by a mesh penetrable for fungal hyphae, but not for roots. Plants were exposed to a C-CO-labeled atmosphere, while N-labeled ammonium and amino acids were added to one side of the split-root system. We found a rapid transfer of recent photosynthates via ectomycorrhizal hyphae to bacteria in root-distant soil areas. Fungal and bacterial phospholipid fatty acid (PLFA) biomarkers were significantly enriched in hyphae-exclusive compartments 24 h after C-CO-labeling. Isotope imaging with nanometer-scale secondary ion mass spectrometry (NanoSIMS) allowed for the first time visualization of plant-derived C and N taken up by an extraradical fungal hypha, and in microbial cells thriving on hyphal surfaces. When N was added to the litter compartments, bacterial biomass, and the amount of incorporated C strongly declined. Interestingly, this effect was also observed in adjacent soil compartments where added N was only available for bacteria through hyphal transport, indicating that ectomycorrhizal fungi were acting on soil bacteria. Together, our results demonstrate that (i) ectomycorrhizal hyphae rapidly transfer plant-derived C to bacterial communities in root-distant areas, and (ii) this transfer promptly responds to changing soil nutrient conditions.
Surface-enhanced Raman spectroscopy of microorganisms: limitations and applicability on the single-cell level.
2019 - Analyst, 3: 943-953
Abstract:
Detection and characterization of microorganisms is essential for both clinical diagnostics and environmental studies. An emerging technique to analyse microbes at single-cell resolution is surface-enhanced Raman spectroscopy (surface-enhanced Raman scattering: SERS). Optimised SERS procedures enable fast analytical read-outs with specific molecular information, providing insight into the chemical composition of microbiological samples. Knowledge about the origin of microbial SERS signals and parameter(s) affecting their occurrence, intensity and/or reproducibility is crucial for reliable SERS-based analyses. In this work, we explore the feasibility and limitations of the SERS approach for characterizing microbial cells and investigate the applicability of SERS for single-cell sorting as well as for three-dimensional visualization of microbial communities. Analyses of six different microbial species (an archaeon, two Gram-positive bacteria, three Gram-negative bacteria) showed that for several of these organisms distinct features in their SERS spectra were lacking. As additional confounding factor, the physiological conditions of the cells (as influenced by e.g., storage conditions or deuterium-labelling) were systematically addressed, for which we conclude that the respective SERS signal at the single-cell level is strongly influenced by the metabolic activity of the analysed cells. While this finding complicates the interpretation of SERS data, it may on the other hand enable probing of the metabolic state of individual cells within microbial populations of interest.
Cyanate and urea are substrates for nitrification by Thaumarchaeota in the marine environment.
2019 - Nat Microbiol, 2: 234-243
Abstract:
Ammonia-oxidizing archaea of the phylum Thaumarchaeota are among the most abundant marine microorganisms. These organisms thrive in the oceans despite ammonium being present at low nanomolar concentrations. Some Thaumarchaeota isolates have been shown to utilize urea and cyanate as energy and N sources through intracellular conversion to ammonium. Yet, it is unclear whether patterns observed in culture extend to marine Thaumarchaeota, and whether Thaumarchaeota in the ocean directly utilize urea and cyanate or rely on co-occurring microorganisms to break these substrates down to ammonium. Urea utilization has been reported for marine ammonia-oxidizing communities, but no evidence of cyanate utilization exists for marine ammonia oxidizers. Here, we demonstrate that in the Gulf of Mexico, Thaumarchaeota use urea and cyanate both directly and indirectly as energy and N sources. We observed substantial and linear rates of nitrite production from urea and cyanate additions, which often persisted even when ammonium was added to micromolar concentrations. Furthermore, single-cell analysis revealed that the Thaumarchaeota incorporated ammonium-, urea- and cyanate-derived N at significantly higher rates than most other microorganisms. Yet, no cyanases were detected in thaumarchaeal genomic data from the Gulf of Mexico. Therefore, we tested cyanate utilization in Nitrosopumilus maritimus, which also lacks a canonical cyanase, and showed that cyanate was oxidized to nitrite. Our findings demonstrate that marine Thaumarchaeota can use urea and cyanate as both an energy and N source. On the basis of these results, we hypothesize that urea and cyanate are substrates for ammonia-oxidizing Thaumarchaeota throughout the ocean.
Sulfate is transported at significant rates through the symbiosome membrane and is crucial for nitrogenase biosynthesis.
2019 - Plant Cell Environ., 4: 1180-1189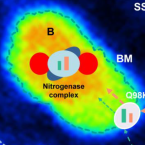
Abstract:
Legume-rhizobia symbioses play a major role in food production for an ever growing human population. In this symbiosis, dinitrogen is reduced ("fixed") to ammonia by the rhizobial nitrogenase enzyme complex and is secreted to the plant host cells, whereas dicarboxylic acids derived from photosynthetically produced sucrose are transported into the symbiosomes and serve as respiratory substrates for the bacteroids. The symbiosome membrane contains high levels of SST1 protein, a sulfate transporter. Sulfate is an essential nutrient for all living organisms, but its importance for symbiotic nitrogen fixation and nodule metabolism has long been underestimated. Using chemical imaging, we demonstrate that the bacteroids take up 20-fold more sulfate than the nodule host cells. Furthermore, we show that nitrogenase biosynthesis relies on high levels of imported sulfate, making sulfur as essential as carbon for the regulation and functioning of symbiotic nitrogen fixation. Our findings thus establish the importance of sulfate and its active transport for the plant-microbe interaction that is most relevant for agriculture and soil fertility.
Microbial nitrogen limitation in the mammalian large intestine.
2018 - Nat Microbiol, 12: 1441-1450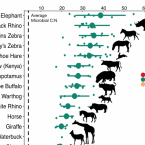
Abstract:
Resource limitation is a fundamental factor governing the composition and function of ecological communities. However, the role of resource supply in structuring the intestinal microbiome has not been established and represents a challenge for mammals that rely on microbial symbionts for digestion: too little supply might starve the microbiome while too much might starve the host. We present evidence that microbiota occupy a habitat that is limited in total nitrogen supply within the large intestines of 30 mammal species. Lowering dietary protein levels in mice reduced their faecal concentrations of bacteria. A gradient of stoichiometry along the length of the gut was consistent with the hypothesis that intestinal nitrogen limitation results from host absorption of dietary nutrients. Nitrogen availability is also likely to be shaped by host-microbe interactions: levels of host-secreted nitrogen were altered in germ-free mice and when bacterial loads were reduced via experimental antibiotic treatment. Single-cell spectrometry revealed that members of the phylum Bacteroidetes consumed nitrogen in the large intestine more readily than other commensal taxa did. Our findings support a model where nitrogen limitation arises from preferential host use of dietary nutrients. We speculate that this resource limitation could enable hosts to regulate microbial communities in the large intestine. Commensal microbiota may have adapted to nitrogen-limited settings, suggesting one reason why excess dietary protein has been associated with degraded gut-microbial ecosystems.
Biodegradation of synthetic polymers in soils: Tracking carbon into CO2 and microbial biomass
2018 - Science Advances, 4: eaas9024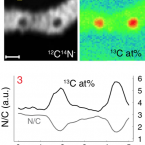
Abstract:
Plastic materials are widely used in agricultural applications to achieve food security for the growing world population. The use of biodegradable instead of nonbiodegradable polymers in single-use agricultural applications, including plastic mulching, promises to reduce plastic accumulation in the environment. We present a novel approach that allows tracking of carbon from biodegradable polymers into CO2 and microbial biomass. The approach is based on 13C-labeled polymers and on isotope-specific analytical methods, including nanoscale secondary ion mass spectrometry (NanoSIMS). Our results unequivocally demonstrate the biodegradability of poly(butylene adipate-co-terephthalate) (PBAT), an important polyester used in agriculture, in soil. Carbon from each monomer unit of PBAT was used by soil microorganisms, including filamentous fungi, to gain energy and to form biomass. This work advances both our conceptual understanding of polymer biodegradation and the methodological capabilities to assess this process in natural and engineered environments.
Ammonia Monooxygenase-Mediated Cometabolic Biotransformation and Hydroxylamine-Mediated Abiotic Transformation of Micropollutants in an AOB/NOB Coculture.
2018 - Environ. Sci. Technol., 16: 9196-9205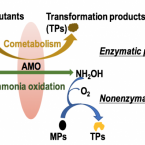
Abstract:
Biotransformation of various micropollutants (MPs) has been found to be positively correlated with nitrification in activated sludge communities. To further elucidate the roles played by ammonia-oxidizing bacteria (AOB) and nitrite-oxidizing bacteria (NOB), we investigated the biotransformation capabilities of an NOB pure culture ( Nitrobacter sp.) and an AOB ( Nitrosomonas europaea)/NOB ( Nitrobacter sp.) coculture for 15 MPs, whose biotransformation was reported previously to be associated with nitrification. The NOB pure culture did not biotransform any investigated MP, whereas the AOB/NOB coculture was capable of biotransforming six MPs (i.e., asulam, bezafibrate, fenhexamid, furosemide, indomethacin, and rufinamide). Transformation products (TPs) were identified, and tentative structures were proposed. Inhibition studies with octyne, an ammonia monooxygenase (AMO) inhibitor, suggested that AMO was the responsible enzyme for MP transformation that occurred cometabolically. For the first time, hydroxylamine, a key intermediate of all aerobic ammonia oxidizers, was found to react with several MPs at concentrations typically occurring in AOB batch cultures. All of these MPs were also biotransformed by the AOB/NOB coculture. Moreover, the same asulam TPs were detected in both biotransformation and hydroxylamine-treated abiotic transformation experiments, whereas rufinamide TPs formed from biological transformation were not detected during hydroxylamine-mediated abiotic transformation, which was consistent with the inability of rufinamide abiotic transformation by hydroxylamine. Thus, in addition to cometabolism likely carried out by AMO, an abiotic transformation route indirectly mediated by AMO might also contribute to MP biotransformation by AOB.
Characterization of the first “Candidatus Nitrotoga” isolate reveals metabolic versatility and separate evolution of widespread nitrite-oxidizing bacteria
2018 - mBio, 9: e01186-18
Abstract:
Nitrification is a key process of the biogeochemical nitrogen cycle and of biological wastewater treatment. The second step, nitrite oxidation to nitrate, is catalyzed by phylogenetically diverse, chemolithoautotrophic nitrite-oxidizing bacteria (NOB). Uncultured NOB from the genus “Candidatus Nitrotoga” are widespread in natural and engineered ecosystems. Knowledge about their biology is sparse, because no genomic information and no pure “Ca. Nitrotoga” culture was available. Here we obtained the first “Ca. Nitrotoga” isolate from activated sludge. This organism, “Candidatus Nitrotoga fabula,” prefers higher temperatures (>20°C; optimum, 24 to 28°C) than previous “Ca. Nitrotoga” enrichments, which were described as cold-adapted NOB. “Ca. Nitrotoga fabula” also showed an unusually high tolerance to nitrite (activity at 30 mM NO2−) and nitrate (up to 25 mM NO3−). Nitrite oxidation followed Michaelis-Menten kinetics, with an apparent Km (Km(app)) of ~89 µM nitrite and a Vmax of ~28 µmol of nitrite per mg of protein per h. Key metabolic pathways of “Ca. Nitrotoga fabula” were reconstructed from the closed genome. “Ca. Nitrotoga fabula” possesses a new type of periplasmic nitrite oxidoreductase belonging to a lineage of mostly uncharacterized proteins. This novel enzyme indicates (i) separate evolution of nitrite oxidation in “Ca. Nitrotoga” and other NOB, (ii) the possible existence of phylogenetically diverse, unrecognized NOB, and (iii) together with new metagenomic data, the potential existence of nitrite-oxidizing archaea. For carbon fixation, “Ca. Nitrotoga fabula” uses the Calvin-Benson-Bassham cycle. It also carries genes encoding complete pathways for hydrogen and sulfite oxidation, suggesting that alternative energy metabolisms enable “Ca. Nitrotoga fabula” to survive nitrite depletion and colonize new niches.
Long-distance electron transport in individual, living cable bacteria.
2018 - Proc. Natl. Acad. Sci. U.S.A., 22: 5786-5791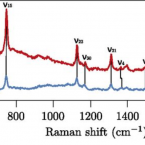
Abstract:
Electron transport within living cells is essential for energy conservation in all respiring and photosynthetic organisms. While a few bacteria transport electrons over micrometer distances to their surroundings, filaments of cable bacteria are hypothesized to conduct electric currents over centimeter distances. We used resonance Raman microscopy to analyze cytochrome redox states in living cable bacteria. Cable-bacteria filaments were placed in microscope chambers with sulfide as electron source and oxygen as electron sink at opposite ends. Along individual filaments a gradient in cytochrome redox potential was detected, which immediately broke down upon removal of oxygen or laser cutting of the filaments. Without access to oxygen, a rapid shift toward more reduced cytochromes was observed, as electrons were no longer drained from the filament but accumulated in the cellular cytochromes. These results provide direct evidence for long-distance electron transport in living multicellular bacteria.
Cultivation and genomic analysis of “Candidatus Nitrosocaldus islandicus”, an obligately thermophilic, ammonia-oxidizing thaumarchaeon from a hot spring biofilm in Graendalur valley, Iceland
2018 - Front Microbiol, 9: 193
Abstract:
Ammonia-oxidizing archaea (AOA) within the phylum Thaumarchaeota are the only known aerobic ammonia oxidizers in geothermal environments. Although molecular data indicate the presence of phylogenetically diverse AOA from the Nitrosocaldus clade, group 1.1b and group 1.1a Thaumarchaeota in terrestrial high-temperature habitats, only one enrichment culture of an AOA thriving above 50 °C has been reported and functionally analyzed. In this study, we physiologically and genomically characterized a newly discovered thaumarchaeon from the deep-branching Nitrosocaldaceae family of which we have obtained a high (~85 %) enrichment from biofilm of an Icelandic hot spring (73 °C). This AOA, which we provisionally refer to as “Candidatus Nitrosocaldus islandicus”, is an obligately thermophilic, aerobic chemolithoautotrophic ammonia oxidizer, which stoichiometricall converts ammonia to nitrite at temperatures between 50 °C and 70 °C. “Ca. N. islandicus” encodes the expected repertoire of enzymes proposed to be required for archaeal ammonia oxidation, but unexpectedly lacks a nirK gene and also possesses no identifiable other enzyme for nitric oxide (NO) generation*. Nevertheless, ammonia oxidation by this AOA appears to be NO-dependent as “Ca. N. islandicus” is, like all other tested AOA, inhibited by the addition of an NO scavenger. Furthermore, comparative genomics revealed that “Ca. N. islandicus” has the potential for aromatic amino acid fermentation as its genome encodes an indolepyruvate oxidoreductase (iorAB) as well as a type 3b hydrogenase, which are not present in any other sequenced AOA. A further surprising genomic feature of this thermophilic ammonia oxidizer is the absence of DNA polymerase D genes – one of the predominant replicative DNA polymerases in all other ammonia-oxidizing Thaumarchaeota. Collectively, our findings suggest that metabolic versatility and DNA replication might differ substantially between obligately thermophilic and other AOA.
Draft genome sequence of Telmatospirillum siberiense 26-4b1T, an acidotolerant peatland alphaproteobacterium potentially involved in sulfur cycling
2018 - Genome Announc, 6: e01524-17
Abstract:
The facultative anaerobic chemoorganoheterotrophic alphaproteobacterium Telmatospirillum siberiense 26-4b1T was isolated from a Siberian peatland. We report on a 6.20 Mbp near complete, high quality draft genome of T. siberiense that reveals expected and novel metabolic potential for the genus Telmatospirillum, including genes for sulfur oxidation.
NanoSIMS and tissue autoradiography reveal symbiont carbon fixation and organic carbon transfer to giant ciliate host.
2018 - ISME J, 3: 714-727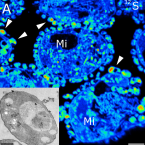
Abstract:
The giant colonial ciliate Zoothamnium niveum harbors a monolayer of the gammaproteobacteria Cand. Thiobios zoothamnicoli on its outer surface. Cultivation experiments revealed maximal growth and survival under steady flow of high oxygen and low sulfide concentrations. We aimed at directly demonstrating the sulfur-oxidizing, chemoautotrophic nature of the symbionts and at investigating putative carbon transfer from the symbiont to the ciliate host. We performed pulse-chase incubations with C- and C-labeled bicarbonate under varying environmental conditions. A combination of tissue autoradiography and nanoscale secondary ion mass spectrometry coupled with transmission electron microscopy was used to follow the fate of the radioactive and stable isotopes of carbon, respectively. We show that symbiont cells fix substantial amounts of inorganic carbon in the presence of sulfide, but also (to a lesser degree) in the absence of sulfide by utilizing internally stored sulfur. Isotope labeling patterns point to translocation of organic carbon to the host through both release of these compounds and digestion of symbiont cells. The latter mechanism is also supported by ultracytochemical detection of acid phosphatase in lysosomes and in food vacuoles of ciliate cells. Fluorescence in situ hybridization of freshly collected ciliates revealed that the vast majority of ingested microbial cells were ectosymbionts.
Ammonia-oxidising archaea living at low pH: Insights from comparative genomics.
2017 - Environ. Microbiol., 12: 4939-4952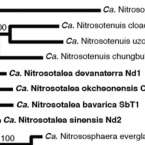
Abstract:
Obligate acidophilic members of the thaumarchaeotal genus Candidatus Nitrosotalea play an important role in nitrification in acidic soils, but their evolutionary and physiological adaptations to acidic environments are still poorly understood, with only a single member of this genus (Ca. N. devanaterra) having its genome sequenced. In this study, we sequenced the genomes of two additional cultured Ca. Nitrosotalea strains, extracted an almost complete Ca. Nitrosotalea metagenome-assembled genome from an acidic fen, and performed comparative genomics of the four Ca. Nitrosotalea genomes with 19 other archaeal ammonia oxidiser genomes. Average nucleotide and amino acid identities revealed that the four Ca. Nitrosotalea strains represent separate species within the genus. The four Ca. Nitrosotalea genomes contained a core set of 103 orthologous gene families absent from all other ammonia-oxidizing archaea and, for most of these gene families, expression could be demonstrated in laboratory culture or the environment via proteomic or metatranscriptomic analyses respectively. Phylogenetic analyses indicated that four of these core gene families were acquired by the Ca. Nitrosotalea common ancestor via horizontal gene transfer from acidophilic representatives of Euryarchaeota. We hypothesize that gene exchange with these acidophiles contributed to the competitive success of the Ca. Nitrosotalea lineage in acidic environments.
Abiotic Conversion of Extracellular NH2OH Contributes to N2O Emission during Ammonia Oxidation.
2017 - Environ. Sci. Technol., 22: 13122-13132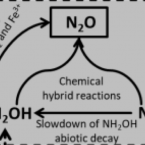
Abstract:
Abiotic processes involving the reactive ammonia-oxidation intermediates nitric oxide (NO) or hydroxylamine (NH2OH) for N2O production have been indicated recently. The latter process would require the availability of substantial amounts of free NH2OH for chemical reactions during ammonia (NH3) oxidation, but little is known about extracellular NH2OH formation by the different clades of ammonia-oxidizing microbes. Here we determined extracellular NH2OH concentrations in culture media of several ammonia-oxidizing bacteria (AOB) and archaea (AOA), as well as one complete ammonia oxidizer (comammox) enrichment (Ca. Nitrospira inopinata) during incubation under standard cultivation conditions. NH2OH was measurable in the incubation media of Nitrosomonas europaea, Nitrosospira multiformis, Nitrososphaera gargensis, and Ca. Nitrosotenuis uzonensis, but not in media of the other tested AOB and AOA. NH2OH was also formed by the comammox enrichment during NH3 oxidation. This enrichment exhibited the largest NH2OH:final product ratio (1.92%), followed by N. multiformis (0.56%) and N. gargensis (0.46%). The maximum proportions of NH4+ converted to N2O via extracellular NH2OH during incubation, estimated on the basis of NH2OH abiotic conversion rates, were 0.12%, 0.08%, and 0.14% for AOB, AOA, and Ca. Nitrospira inopinata, respectively, and were consistent with published NH4+:N2O conversion ratios for AOB and AOA.
Kinetic analysis of a complete nitrifier reveals an oligotrophic lifestyle.
2017 - Nature, 549: 269-272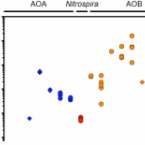
Abstract:
Nitrification, the oxidation of ammonia (NH3) via nitrite (NO2(-)) to nitrate (NO3(-)), is a key process of the biogeochemical nitrogen cycle. For decades, ammonia and nitrite oxidation were thought to be separately catalysed by ammonia-oxidizing bacteria (AOB) and archaea (AOA), and by nitrite-oxidizing bacteria (NOB). The recent discovery of complete ammonia oxidizers (comammox) in the NOB genus Nitrospira, which alone convert ammonia to nitrate, raised questions about the ecological niches in which comammox Nitrospira successfully compete with canonical nitrifiers. Here we isolate a pure culture of a comammox bacterium, Nitrospira inopinata, and show that it is adapted to slow growth in oligotrophic and dynamic habitats on the basis of a high affinity for ammonia, low maximum rate of ammonia oxidation, high growth yield compared to canonical nitrifiers, and genomic potential for alternative metabolisms. The nitrification kinetics of four AOA from soil and hot springs were determined for comparison. Their surprisingly poor substrate affinities and lower growth yields reveal that, in contrast to earlier assumptions, AOA are not necessarily the most competitive ammonia oxidizers present in strongly oligotrophic environments and that N. inopinata has the highest substrate affinity of all analysed ammonia oxidizer isolates except the marine AOA Nitrosopumilus maritimus SCM1 (ref. 3). These results suggest a role for comammox organisms in nitrification under oligotrophic and dynamic conditions.
AmoA-targeted polymerase chain reaction primers for the specific detection and quantification of comammox Nitrospira in the environment
2017 - Front Microbiol, 8:1508
Abstract:
Nitrification, the oxidation of ammonia via nitrite to nitrate, has always been considered to be catalyzed by the concerted activity of ammonia- and nitrite-oxidizing microorganisms. Only recently, complete ammonia oxidizers (‘comammox’), which oxidize ammonia to nitrate on their own, were identified in the bacterial genus Nitrospira, previously assumed to contain only canonical nitrite oxidizers. Nitrospira are widespread in nature, but for assessments of the distribution and functional importance of comammox Nitrospira in ecosystems, cultivation-independent tools to distinguish comammox from strictly nitrite oxidizing Nitrospira are required. Here we developed new PCR primer sets that specifically target the amoA genes coding for subunit A of the distinct ammonia monooxygenase of comammox Nitrospira. While existing primers capture only a fraction of the known comammox amoA diversity, the new primer sets cover as much as 95% of the comammox amoA clade A and 92% of the clade B sequences in a reference database containing 326 comammox amoA genes with sequence information at the primer binding sites. Application of the primers to 13 samples from engineered systems (a groundwater well, drinking water treatment and wastewater treatment plants) and other habitats (rice paddy and forest soils, rice rhizosphere, brackish lake sediment and freshwater biofilm) detected comammox Nitrospira in all samples and revealed a considerable diversity of comammox in most habitats. Excellent primer specificity for comammox amoA was achieved by avoiding the use of highly degenerate primer preparations and by using equimolar mixtures of oligonucleotides that match existing comammox amoA genes. Quantitative PCR with these equimolar primer mixtures was highly sensitive and specific, and enabled the efficient quantification of clade A and clade B comammox amoA gene copy numbers in environmental samples. The measured relative abundances of comammox Nitrospira, compared to canonical ammonia oxidizers, were highly variable across environments. The new comammox amoA-targeted primers enable more encompassing future studies of nitrifying microorganisms in diverse habitats. For example, they may be used to monitor the population dynamics of uncultured comammox organisms under changing environmental conditions and in response to altered treatments in engineered and agricultural ecosystems.
Giant viruses with an expanded complement of translation system components.
2017 - Science, 6333: 82-85
Abstract:
The discovery of giant viruses blurred the sharp division between viruses and cellular life. Giant virus genomes encode proteins considered as signatures of cellular organisms, particularly translation system components, prompting hypotheses that these viruses derived from a fourth domain of cellular life. Here we report the discovery of a group of giant viruses (Klosneuviruses) in metagenomic data. Compared with other giant viruses, the Klosneuviruses encode an expanded translation machinery, including aminoacyl transfer RNA synthetases with specificities for all 20 amino acids. Notwithstanding the prevalence of translation system components, comprehensive phylogenomic analysis of these genes indicates that Klosneuviruses did not evolve from a cellular ancestor but rather are derived from a much smaller virus through extensive gain of host genes.
Crenothrix are major methane consumers in stratified lakes.
2017 - ISME J, 9: 2124-2140
Abstract:
Methane-oxidizing bacteria represent a major biological sink for methane and are thus Earth's natural protection against this potent greenhouse gas. Here we show that in two stratified freshwater lakes a substantial part of upward-diffusing methane was oxidized by filamentous gamma-proteobacteria related to Crenothrix polyspora. These filamentous bacteria have been known as contaminants of drinking water supplies since 1870, but their role in the environmental methane removal has remained unclear. While oxidizing methane, these organisms were assigned an 'unusual' methane monooxygenase (MMO), which was only distantly related to 'classical' MMO of gamma-proteobacterial methanotrophs. We now correct this assignment and show that Crenothrix encode a typical gamma-proteobacterial PmoA. Stable isotope labeling in combination swith single-cell imaging mass spectrometry revealed methane-dependent growth of the lacustrine Crenothrix with oxygen as well as under oxygen-deficient conditions. Crenothrix genomes encoded pathways for the respiration of oxygen as well as for the reduction of nitrate to N2O. The observed abundance and planktonic growth of Crenothrix suggest that these methanotrophs can act as a relevant biological sink for methane in stratified lakes and should be considered in the context of environmental removal of methane.
Capturing the genetic makeup of the active microbiome in situ.
2017 - ISME J, 9: 1949-1963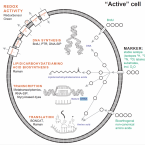
Abstract:
More than any other technology, nucleic acid sequencing has enabled microbial ecology studies to be complemented with the data volumes necessary to capture the extent of microbial diversity and dynamics in a wide range of environments. In order to truly understand and predict environmental processes, however, the distinction between active, inactive and dead microbial cells is critical. Also, experimental designs need to be sensitive toward varying population complexity and activity, and temporal as well as spatial scales of process rates. There are a number of approaches, including single-cell techniques, which were designed to study in situ microbial activity and that have been successively coupled to nucleic acid sequencing. The exciting new discoveries regarding in situ microbial activity provide evidence that future microbial ecology studies will indispensably rely on techniques that specifically capture members of the microbiome active in the environment. Herein, we review those currently used activity-based approaches that can be directly linked to shotgun nucleic acid sequencing, evaluate their relevance to ecology studies, and discuss future directions.
Cultivation and characterization of Candidatus Nitrosocosmicus exaquare, an ammonia-oxidizing archaeon from a municipal wastewater treatment system.
2017 - ISME J, 5: 1142-1157
Abstract:
Thaumarchaeota have been detected in several industrial and municipal wastewater treatment plants (WWTPs), despite the fact that ammonia-oxidizing archaea (AOA) are thought to be adapted to low ammonia environments. However, the activity, physiology and metabolism of WWTP-associated AOA remain poorly understood. We report the cultivation and complete genome sequence of Candidatus Nitrosocosmicus exaquare, a novel AOA representative from a municipal WWTP in Guelph, Ontario (Canada). In enrichment culture, Ca. N. exaquare oxidizes ammonia to nitrite stoichiometrically, is mesophilic, and tolerates at least 15 mm of ammonium chloride or sodium nitrite. Microautoradiography (MAR) for enrichment cultures demonstrates that Ca. N. exaquare assimilates bicarbonate in association with ammonia oxidation. However, despite using inorganic carbon, the ammonia-oxidizing activity of Ca. N. exaquare is greatly stimulated in enrichment culture by the addition of organic compounds, especially malate and succinate. Ca. N. exaquare cells are coccoid with a diameter of ~1-2 μm. Phylogenetically, Ca. N. exaquare belongs to the Nitrososphaera sister cluster within the Group I.1b Thaumarchaeota, a lineage which includes most other reported AOA sequences from municipal and industrial WWTPs. The 2.99 Mbp genome of Ca. N. exaquare encodes pathways for ammonia oxidation, bicarbonate fixation, and urea transport and breakdown. In addition, this genome encodes several key genes for dealing with oxidative stress, including peroxidase and catalase. Incubations of WWTP biofilm demonstrate partial inhibition of ammonia-oxidizing activity by 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), suggesting that Ca. N. exaquare-like AOA may contribute to nitrification in situ. However, CARD-FISH-MAR showed no incorporation of bicarbonate by detected Thaumarchaeaota, suggesting that detected AOA may incorporate non-bicarbonate carbon sources or rely on an alternative and yet unknown metabolism.
A New Perspective on Microbes Formerly Known as Nitrite-Oxidizing Bacteria.
2016 - Trends Microbiol., 9: 699-712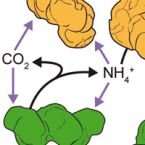
Abstract:
Nitrite-oxidizing bacteria (NOB) catalyze the second step of nitrification, nitrite oxidation to nitrate, which is an important process of the biogeochemical nitrogen cycle. NOB were traditionally perceived as physiologically restricted organisms and were less intensively studied than other nitrogen-cycling microorganisms. This picture is in contrast to new discoveries of an unexpected high diversity of mostly uncultured NOB and a great physiological versatility, which includes complex microbe-microbe interactions and lifestyles outside the nitrogen cycle. Most surprisingly, close relatives to NOB perform complete nitrification (ammonia oxidation to nitrate) and this finding will have far-reaching implications for nitrification research. We review recent work that has changed our perspective on NOB and provides a new basis for future studies on these enigmatic organisms.
Single cell stable isotope probing in microbiology using Raman microspectroscopy
2016 - Curr Opin Biotechnol, 41:34-42
Abstract:
Microbial communities are essential for most ecosystem processes and interact in highly complex ways with virtually all eukaryotes. Thus, a detailed understanding of the function of such communities is a fundamental prerequisite for microbial ecologists, applied microbiologists and microbiome researchers. Using single cell Raman microspectroscopy, biochemical fingerprints of individual microbial cells can be obtained in a fast and nondestructive manner. If combined with stable isotope probing (SIP), Raman spectroscopy can directly reveal functions of single microorganisms in their natural habitat. This review provides an update on various SIP-approaches suitable for combination with different Raman scattering techniques and illustrates how single cell Raman SIP can be directly combined with the omics-centric analysis pipelines generally applied to investigate microbial communities.
Biotransformation of Two Pharmaceuticals by the Ammonia-Oxidizing Archaeon Nitrososphaera gargensis.
2016 - Environ Sci Technol, 9: 4682-92
Abstract:
The biotransformation of some micropollutants has previously been observed to be positively associated with ammonia oxidation activities and the transcript abundance of the archaeal ammonia monooxygenase gene (amoA) in nitrifying activated sludge. Given the increasing interest in and potential importance of ammonia-oxidizing archaea (AOA), we investigated the capabilities of an AOA pure culture, Nitrososphaera gargensis, to biotransform ten micropollutants belonging to three structurally similar groups (i.e., phenylureas, tertiary amides, and tertiary amines). N. gargensis was able to biotransform two of the tertiary amines, mianserin (MIA) and ranitidine (RAN), exhibiting similar compound specificity as two ammonia-oxidizing bacteria (AOB) strains that were tested for comparison. The same MIA and RAN biotransformation reactions were carried out by both the AOA and AOB strains. The major transformation product (TP) of MIA, α-oxo MIA was likely formed via a two-step oxidation reaction. The first hydroxylation step is typically catalyzed by monooxygenases. Three RAN TP candidates were identified from nontarget analysis. Their tentative structures and possible biotransformation pathways were proposed. The biotransformation of MIA and RAN only occurred when ammonia oxidation was active, suggesting cometabolic transformations. Consistently, a comparative proteomic analysis revealed no significant differential expression of any protein-encoding gene in N. gargensis grown on ammonium with MIA or RAN compared with standard cultivation on ammonium only. Taken together, this study provides first important insights regarding the roles played by AOA in micropollutant biotransformation.
Multi-scale imaging of anticancer platinum(IV) compounds in murine tumor and kidney
2016 - Chemical Science, 7: 3052-3061
Abstract:
Nano-scale secondary ion mass spectrometry (NanoSIMS) enables trace element and isotope analyses with high spatial resolution. This unique capability has recently been exploited in several studies analyzing the subcellular distribution of Au and Pt anticancer compounds. However, these studies were restricted to cell culture systems. To explore the applicability to the in vivo setting, we developed a combined imaging approach consisting of laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), NanoSIMS and transmission electron microscopy (TEM) suitable for multi-scale detection of the platinum distribution in tissues. Applying this approach to murine tumor and kidney samples upon administration of selected platinum(IV) anticancer prodrugs revealed uneven platinum distributions on both the organ and subcellular scales. Spatial platinum accumulation patterns by LA-ICP-MS were quantitatively assessed in histologically heterogeneous organs (e.g., higher platinum accumulation in kidney cortex than in medulla) and used to select regions of interest for subcellular scale imaging with NanoSIMS. These analyses revealed cytoplasmic sulfur-rich organelles to accumulate platinum in both kidney and malignant cells. Those in the tumor were subsequently identified as organelles of lysosomal origin, demonstrating the potential of the combinatorial approach for investigating therapeutically relevant drug concentrations on a submicrometer scale.
Ecophysiology of an uncultivated lineage of Aigarchaeota from an oxic, hot spring filamentous 'streamer' community.
2016 - ISME J, 1: 210-224
Abstract:
The candidate archaeal phylum 'Aigarchaeota' contains microorganisms from terrestrial and subsurface geothermal ecosystems. The phylogeny and metabolic potential of Aigarchaeota has been deduced from several recent single-cell amplified genomes; however, a detailed description of their metabolic potential and in situ transcriptional activity is absent. Here, we report a comprehensive metatranscriptome-based reconstruction of the in situ metabolism of Aigarchaeota in an oxic, hot spring filamentous 'streamer' community. Fluorescence in situ hybridization showed that these newly discovered Aigarchaeota are filamentous, which is consistent with the presence and transcription of an actin-encoding gene. Aigarchaeota filaments are intricately associated with other community members, which include both bacteria (for example, filamentous Thermocrinis spp.) and archaea. Metabolic reconstruction of genomic and metatranscriptomic data suggests that this aigarchaeon is an aerobic, chemoorganoheterotroph with autotrophic potential. A heme copper oxidase complex was identified in the environmental genome assembly and highly transcribed in situ. Potential electron donors include acetate, fatty acids, amino acids, sugars and aromatic compounds, which may originate from extracellular polymeric substances produced by other microorganisms shown to exist in close proximity and/or autochthonous dissolved organic carbon (OC). Transcripts related to genes specific to each of these potential electron donors were identified, indicating that this aigarchaeon likely utilizes several OC substrates. Characterized members of this lineage cannot synthesize heme, and other cofactors and vitamins de novo, which suggests auxotrophy. We propose the name Candidatus 'Calditenuis aerorheumensis' for this aigarchaeon, which describes its filamentous morphology and its primary electron acceptor, oxygen.
Intestinal microbiota signatures associated with inflammation history in mice experiencing recurring colitis
2015 - Front Microbiol, 6: 1408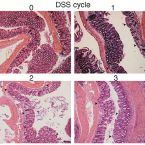
Abstract:
Acute colitis causes alterations in the intestinal microbiota, but the microbiota is thought to recover after such events. Extreme microbiota alterations are characteristic of human chronic inflammatory bowel diseases, although alterations reported in different studies are divergent and sometimes even contradictory. To better understand the impact of periodic disturbances on the intestinal microbiota and its compositional difference between acute and relapsing colitis, we investigated the beginnings of recurrent inflammation using the dextran sodium sulfate (DSS) mouse model of chemically induced colitis. Using bacterial 16S rRNA gene-targeted pyrosequencing as well as quantitative fluorescence in situ hybridization, we profiled the intestinal and stool microbiota of mice over the course of three rounds of DSS-induced colitis and recovery. We found that characteristic inflammation-associated microbiota could be detected in recovery-phase mice. Successive inflammation episodes further drove the microbiota into an increasingly altered composition post-inflammation, and signatures of colitis history were detectable in the microbiota more sensitively than by pathology analysis. Bacterial indicators of murine colitis history were identified in intestinal and stool samples, with a high degree of consistency between both sample types. Stool may therefore be a promising non-invasive source of bacterial biomarkers that are highly sensitive to inflammation state and history.
Complete nitrification by Nitrospira bacteria
2015 - Nature, 528: 504-509
Abstract:
Nitrification, the oxidation of ammonia via nitrite to nitrate, has always been considered to be a two-step process catalysed by chemolithoautotrophic microorganisms oxidizing either ammonia or nitrite. No known nitrifier carries out both steps, although complete nitrification should be energetically advantageous. This functional separation has puzzled microbiologists for a century. Here we report on the discovery and cultivation of a completely nitrifying bacterium from the genus Nitrospira, a globally distributed group of nitrite oxidizers. The genome of this chemolithoautotrophic organism encodes the pathways both for ammonia and nitrite oxidation, which are concomitantly activated during growth by ammonia oxidation to nitrate. Genes affiliated with the phylogenetically distinct ammonia monooxygenase and hydroxylamine dehydrogenase genes of Nitrospira are present in many environments and were retrieved on Nitrospira-contigs in new metagenomes from engineered systems. These findings fundamentally change our picture of nitrification and point to completely nitrifying Nitrospira as key components of nitrogen-cycling microbial communities.
Conductive consortia
2015 - Nature News & Views, 526: 513-514
Abstract:
Physiological analyses, electron microscopy and single-cell chemical imaging suggest that direct electron transfer occurs between the members of methane-oxidizing microbial consortia.
Advancements in the application of NanoSIMS and Raman microspectroscopy to investigate the activity of microbial cells in soils
2015 - FEMS Microbiology Ecology - *Editor's Choice Article*, in press
Abstract:
The combined approach of incubating environmental samples with stable isotope-labeled substrates followed by single-cell analyses through high-resolution secondary ion mass spectrometry (NanoSIMS) or Raman microspectroscopy provides insights into the in situ function of microorganisms. This approach has found limited application in soils presumably due to the dispersal of microbial cells in a large background of particles. We developed a pipeline for the efficient preparation of cell extracts from soils for subsequent single-cell methods by combining cell detachment with separation of cells and soil particles followed by cell concentration. The procedure was evaluated by examining its influence on cell recoveries and microbial community composition across two soils. This approach generated a cell fraction with considerably reduced soil particle load and of sufficient small size to allow single-cell analysis by NanoSIMS, as shown when detecting active N2-fixing and cellulose-responsive microorganisms via 15N2 and 13C-UL-cellulose incubations, respectively. The same procedure was also applicable for Raman microspectroscopic analyses of soil microorganisms, assessed via microcosm incubations with a 13C-labeled carbon source and deuterium oxide (D2O, a general activity marker). The described sample preparation procedure enables single-cell analysis of soil microorganisms using NanoSIMS and Raman microspectroscopy, but should also facilitate single-cell sorting and sequencing.
Intestinal epithelial cell tyrosine kinase 2 transduces interleukin-22 signals to protect from acute colitis
2015 - J Immunol., 195: 5011-5024
Abstract:
In the intestinal tract, IL-22 activates signal transducer and activator of transcription 3 (STAT3) to promote intestinal epithelial cell (IEC) homeostasis and tissue healing. The mechanism has remained obscure but we demonstrate that IL-22 acts via tyrosine kinase 2 (Tyk2), a member of the Janus kinase (Jak) family. Using a mouse model for colitis, we show that Tyk2 deficiency is associated with an altered composition of the gut microbiota and exacerbates inflammatory bowel disease (IBD). Colitic Tyk2-/- mice have less phosphorylated STAT3 (pY-STAT3) in colon tissue and their IECs proliferate less efficiently. Tyk2-deficient primary IECs show reduced pY-STAT3 in response to IL-22 stimulation and expression of IL-22-STAT3 target genes is reduced in IECs from healthy and colitic Tyk2-/- mice. Experiments with conditional Tyk2-/- mice reveal that IEC-specific depletion of Tyk2 aggravates colitis. Disease symptoms can be alleviated by administering high doses of recombinant IL-22-Fc, indicating that Tyk2 deficiency can be rescued via the IL-22 receptor complex. The pivotal function of Tyk2 in IL-22-dependent colitis was confirmed in Citrobacter rodentium-induced disease. Thus, Tyk2 protects against acute colitis in part by amplifying inflammation-induced epithelial IL-22 signaling to STAT3.
Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira
2015 - Proc Natl Acad Sci U S A, 112: 11371-11376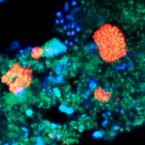
Abstract:
Nitrospira are a diverse group of nitrite-oxidizing bacteria and among the environmentally most widespread nitrifiers. Despite this, they remain scarcely studied and mostly uncultured. Based on genomic and experimental data from Nitrospira moscoviensis representing the ubiquitous Nitrospira lineage II, we identified ecophysiological traits that contribute to the ecological success of Nitrospira. Unexpectedly, N. moscoviensis possesses genes coding for a urease and cleaves urea to ammonia and CO2. Ureolysis was not observed yet in nitrite oxidizers and enables N. moscoviensis to supply ammonia oxidizers lacking urease with ammonia from urea, which is fully nitrified by this consortium through reciprocal feeding. The presence of highly similar urease genes in Nitrospira lenta from activated sludge, in metagenomes from soils and freshwater habitats, and of other ureases in marine nitrite oxidizers, suggests a wide distribution of this extended interaction between ammonia and nitrite oxidizers, which enables nitrite-oxidizing bacteria to indirectly utilize urea as a source of energy. A soluble formate dehydrogenase lends additional ecophysiological flexibility and allows N. moscoviensis to utilize formate, with or without concomitant nitrite oxidation, using oxygen, nitrate, or both compounds as terminal electron acceptors. Compared to Nitrospira defluvii from lineage I, N. moscoviensis shares the Nitrospira core metabolism but shows substantial genomic dissimilarity including genes for adaptations to elevated oxygen concentrations. Reciprocal feeding and metabolic versatility, including the participation in different nitrogen cycling processes, likely are key factors for the niche partitioning, the ubiquity, and the high diversity of Nitrospira in natural and engineered ecosystems.
Endosymbionts escape dead hydrothermal vent tubeworms to enrich the free-living population.
2015 - Proc. Natl. Acad. Sci. U.S.A., 36: 11300-11305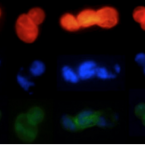
Abstract:
Theory predicts that horizontal acquisition of symbionts by plants and animals must be coupled to release and limited dispersal of symbionts for intergenerational persistence of mutualisms. For deep-sea hydrothermal vent tubeworms (Vestimentifera, Siboglinidae), it has been demonstrated that a few symbiotic bacteria infect aposymbiotic host larvae and grow in a newly formed organ, the trophosome. However, whether viable symbionts can be released to augment environmental populations has been doubtful, because (i) the adult worms lack obvious openings and (ii) the vast majority of symbionts has been regarded as terminally differentiated. Here we show experimentally that symbionts rapidly escape their hosts upon death and recruit to surfaces where they proliferate. Estimating symbiont release from our experiments taken together with well-known tubeworm density ranges, we suggest a few million to 1.5 billion symbionts seeding the environment upon death of a tubeworm clump. In situ observations show that such clumps have rapid turnover, suggesting that release of large numbers of symbionts may ensure effective dispersal to new sites followed by active larval colonization. Moreover, release of symbionts might enable adaptations that evolve within host individuals to spread within host populations and possibly to new environments.
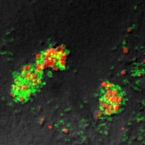
Abstract:
Ammonia- and nitrite-oxidizing microorganisms are collectively responsible for the aerobic oxidation of ammonia via nitrite to nitrate and have essential roles in the global biogeochemical nitrogen cycle. The physiology of nitrifiers has been intensively studied, and urea and ammonia are the only recognized energy sources that promote the aerobic growth of ammonia-oxidizing bacteria and archaea. Here we report the aerobic growth of a pure culture of the ammonia-oxidizing thaumarchaeote Nitrososphaera gargensis using cyanate as the sole source of energy and reductant; to our knowledge, the first organism known to do so. Cyanate, a potentially important source of reduced nitrogen in aquatic and terrestrial ecosystems, is converted to ammonium and carbon dioxide in Nitrososphaera gargensis by a cyanase enzyme that is induced upon addition of this compound. Within the cyanase gene family, this cyanase is a member of a distinct clade also containing cyanases of nitrite-oxidizing bacteria of the genus Nitrospira. We demonstrate by co-culture experiments that these nitrite oxidizers supply cyanase-lacking ammonia oxidizers with ammonium from cyanate, which is fully nitrified by this microbial consortium through reciprocal feeding. By screening a comprehensive set of more than 3,000 publically available metagenomes from environmental samples, we reveal that cyanase-encoding genes clustering with the cyanases of these nitrifiers are widespread in the environment. Our results demonstrate an unexpected metabolic versatility of nitrifying microorganisms, and suggest a previously unrecognized importance of cyanate in cycling of nitrogen compounds in the environment.
Revisiting N₂ fixation in Guerrero Negro intertidal microbial mats with a functional single-cell approach
2015 - ISME J, 9: 485-96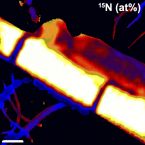
Abstract:
Photosynthetic microbial mats are complex, stratified ecosystems in which high rates of primary production create a demand for nitrogen, met partially by N₂ fixation. Dinitrogenase reductase (nifH) genes and transcripts from Cyanobacteria and heterotrophic bacteria (for example, Deltaproteobacteria) were detected in these mats, yet their contribution to N2 fixation is poorly understood. We used a combined approach of manipulation experiments with inhibitors, nifH sequencing and single-cell isotope analysis to investigate the active diazotrophic community inintertidal microbial mats at Laguna Ojo de Liebre near Guerrero Negro, Mexico. Acetylene reduction assays with specific metabolic inhibitors suggested that both sulfate reducers and members of the Cyanobacteria contributed to N₂ fixation, whereas (15)N₂ tracer experiments at the bulk level only supported a contribution of Cyanobacteria. Cyanobacterial and nifH Cluster III (including deltaproteobacterial sulfate reducers) sequences dominated the nifH gene pool, whereas the nifH transcript pool was dominated by sequences related to Lyngbya spp. Single-cell isotope analysis of (15)N₂-incubated mat samples via high-resolution secondary ion mass spectrometry (NanoSIMS) revealed that Cyanobacteria were enriched in (15)N, with the highest enrichment being detected in Lyngbya spp. filaments (on average 4.4 at% (15)N), whereas the Deltaproteobacteria (identified by CARD-FISH) were not significantly enriched. We investigated the potential dilution effect from CARD-FISH on the isotopic composition and concluded that the dilution bias was not substantial enough to influence our conclusions. Our combined data provide evidence that members of the Cyanobacteria, especially Lyngbya spp., actively contributed to N₂ fixation in the intertidal mats, whereas support for significant N₂ fixation activity of the targeted deltaproteobacterial sulfate reducers could not be found.
Inhibitory properties of C2 - C10 1-alkynes on ammonia oxidation in two Nitrososphaera species
2015 - Appl Environ Microbiol, 81: 1942-8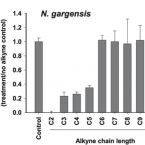
Abstract:
A previous study showed that ammonia oxidation by the Thaumarchaeota Nitrosopumilus maritimus (group 1.1a) was resistant to concentrations of the C8 1-alkyne, octyne, which completely inhibits activity by ammonia-oxidizing bacteria. In this study, the inhibitory effects of octyne and other C2 to C10 1-alkynes were evaluated on the nitrite production activity of two pure culture isolates from Thaumarchaeota group 1.1b, Nitrososphaera viennensis strain EN76 and Nitrososphaera gargensis. Both N. viennensis and N. gargensis were insensitive to concentrations of octyne that cause complete and irreversible inactivation of nitrite production by ammonia-oxidizing bacteria. However, octyne concentrations (≥20 μM) that did not inhibit N. maritimus partially inhibited nitrite production in N. viennensis and N. gargensis in a manner that did not show the characteristics of irreversible inactivation. In contrast to previous studies with an ammonia-oxidizing bacterium, Nitrosomonas europaea, octyne inhibition of N. viennensis was: (i) fully and immediately reversible, (ii) not competitive with NH4 (+), and (iii) without effect on the competitive interaction between NH4 (+) and acetylene. Both N. viennensis and N. gargensis demonstrated the same overall trend in regard to 1-alkyne inhibition as previously observed for N. maritimus, being highly sensitive to ≤C5 alkynes and more resistant to longer-chain length alkynes. Reproducible differences were observed among N. maritimus, N. viennensis, and N. gargensis in regard to the extent of their resistance/sensitivity to C6 and C7 1-alkynes, which may indicate differences in the ammonia monooxygenase binding and catalytic site(s) among the Thaumarchaeota.
A nanoscale secondary ion mass spectrometry study of dinoflagellate functional diversity in reef-building corals.
2015 - Environ. Microbiol., 10: 3570-80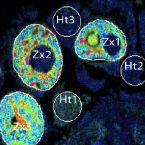
Abstract:
Nutritional interactions between corals and symbiotic dinoflagellate algae lie at the heart of the structural foundation of coral reefs. Whilst the genetic diversity of Symbiodinium has attracted particular interest because of its contribution to the sensitivity of corals to environmental changes and bleaching (i.e. disruption of coral-dinoflagellate symbiosis), very little is known about the in hospite metabolic capabilities of different Symbiodinium types. Using a combination of stable isotopic labelling and nanoscale secondary ion mass spectrometry (NanoSIMS), we investigated the ability of the intact symbiosis between the reef-building coral Isopora palifera, and Symbiodinium C or D types, to assimilate dissolved inorganic carbon (via photosynthesis) and nitrogen (as ammonium). Our results indicate that Symbiodinium types from two clades naturally associated with I. palifera possess different metabolic capabilities. The Symbiodinium C type fixed and passed significantly more carbon and nitrogen to its coral host than the D type. This study provides further insights into the metabolic plasticity among different Symbiodinium types in hospite and strengthens the evidence that the more temperature-tolerant Symbiodinium D type may be less metabolically beneficial for its coral host under non-stressful conditions.
Nitrotoga-like bacteria are previously unrecognized key nitrite oxidizers in full-scale wastewater treatment plants
2015 - ISME J, 9: 708-720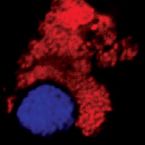
Abstract:
Numerous past studies have shown members of the genus Nitrospira to be the predominant nitrite-oxidizing bacteria (NOB) in nitrifying wastewatertreatment plants (WWTPs). Only recently, the novel NOB 'Candidatus Nitrotoga arctica' was identified in permafrost soil and a close relative was enriched from activated sludge. Still, little is known about diversity, distribution and functional importance of Nitrotoga in natural and engineered ecosystems. Here we developed Nitrotoga 16S rRNA-specific PCR primers and fluorescence in situ hybridization (FISH) probes, which were applied to screen activated sludge samples from 20 full-scale WWTPs. Nitrotoga-like bacteria were detected by PCR in 11 samples and reached abundances detectable by FISH in seven sludges. They coexisted with Nitrospira in most of these WWTPs, but constituted the only detectable NOB in two systems. Quantitative FISH revealed that Nitrotoga accounted for nearly 2% of the total bacterial community in one of these plants, a number comparable to Nitrospira abundances in other WWTPs. Spatial statistics revealed that Nitrotoga coaggregated with ammonia-oxidizing bacteria, strongly supporting a functional role in nitrite oxidation. This activity was confirmed by FISH in combination with microradiography, which revealednitrite-dependent autotrophic carbon fixation by Nitrotoga in situ. Correlation of the presence or absence with WWTP operational parameters indicated low temperatures as a main factor supporting high Nitrotoga abundances, although in incubation experiments these NOB remained active over an unexpected range of temperatures, and also at different ambient nitrite concentrations. In conclusion, this study demonstrates that Nitrotoga can be functionally important nitrite oxidizers in WWTPs and can even represent the only known NOB in engineered systems.
Functionally relevant diversity of closely related Nitrospira in activated sludge
2015 - ISME J, 9: 643-655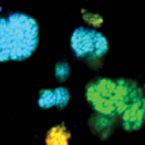
Abstract:
Nitrospira are chemolithoautotrophic nitrite-oxidizing bacteria that catalyze the second step of nitrification in most oxic habitats and are important for excess nitrogen removal from sewage in wastewater treatment plants (WWTPs). To date, little is known about their diversity and ecological niche partitioning within complex communities. In this study, the fine-scale community structure and function of Nitrospira was analyzed in two full-scale WWTPs as model ecosystems. In Nitrospira-specific 16S rRNA clone libraries retrieved from each plant, closely related phylogenetic clusters (16S rRNA identities between clusters ranged from 95.8% to 99.6%) within Nitrospira lineages I and II were found. Newly designed probes for fluorescence in situ hybridization (FISH) allowed the specific detection of several of these clusters, whose coexistence in the WWTPs was shown for prolonged periods of several years. In situ ecophysiological analyses based on FISH, relative abundance and spatial arrangement quantification, as well as microautoradiography revealed functional differences of these Nitrospira clusters regarding the preferred nitrite concentration, the utilization of formate as substrate and the spatial coaggregation with ammonia-oxidizing bacteria as symbiotic partners. Amplicon pyrosequencing of the nxrB gene, which encodes subunit beta of nitrite oxidoreductase of Nitrospira, revealed in one of the WWTPs as many as 121 species-level nxrB operational taxonomic units with highly uneven relative abundances in the amplicon library. These results show a previously unrecognized highdiversity of Nitrospira in engineered systems, which is at least partially linked to niche differentiation and may have important implications for process stability.
Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells
2015 - Proc Natl Acad Sci USA, 112: E194-203
Abstract:
Microbial communities are essential to the function of virtually all ecosystems and eukaryotes, including humans. However, it is still a major challenge to identify microbial cells active under natural conditions in complex systems. In this study, we developed a new method to identify and sort active microbes on the single-cell level in complex samples using stable isotope probing with heavy water (D2O) combined with Raman microspectroscopy. Incorporation of D2O-derived D into the biomass of autotrophic and heterotrophic bacteria and archaea could be unambiguously detected via C-D signature peaks in single-cell Raman spectra, and the obtained labeling pattern was confirmed by nanoscale-resolution secondary ion MS. In fast-growing Escherichia coli cells, label detection was already possible after 20 min. For functional analyses of microbial communities, the detection of D incorporation from D2O in individual microbial cells via Raman microspectroscopy can be directly combined with FISH for the identification of active microbes. Applying this approach to mouse cecal microbiota revealed that the host-compound foragers Akkermansia muciniphila and Bacteroides acidifaciens exhibited distinctive response patterns to amendments of mucin and sugars. By Raman-based cell sortingof active (deuterated) cells with optical tweezers and subsequent multiple displacement amplification and DNA sequencing, novel cecal microbes stimulated by mucin and/or glucosamine were identified, demonstrating the potential of the nondestructive D2O-Raman approach for targeted sortingof microbial cells with defined functional properties for single-cell genomics.
Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains.
2014 - PLoS Biol., 12(8):e1001920
Abstract:
Microbes hold the key to life. They hold the secrets to our past (as the descendants of the earliest forms of life) and the prospects for our future (as we mine their genes for solutions to some of the planet's most pressing problems, from global warming to antibiotic resistance). However, the piecemeal approach that has defined efforts to study microbial genetic diversity for over 20 years and in over 30,000 genome projects risks squandering that promise. These efforts have covered less than 20% of the diversity of the cultured archaeal and bacterial species, which represent just 15% of the overall known prokaryotic diversity. Here we call for the funding of a systematic effort to produce a comprehensive genomic catalog of all cultured Bacteria and Archaea by sequencing, where available, the type strain of each species with a validly published name (currently∼11,000). This effort will provide an unprecedented level of coverage of our planet's genetic diversity, allow for the large-scale discovery of novel genes and functions, and lead to an improved understanding of microbial evolution and function in the environment.
Biology of a widespread uncultivated archaeon that contributes to carbon fixation in the subsurface.
2014 - Nat Commun., 5: 5497
Abstract:
Subsurface microbial life contributes significantly to biogeochemical cycling, yet it remains largely uncharacterized, especially its archaeal members. This 'microbial dark matter' has been explored by recent studies that were, however, mostly based on DNA sequence information only. Here, we use diverse techniques including ultrastuctural analyses to link genomics to biology for the SM1 Euryarchaeon lineage, an uncultivated group ofsubsurface archaea. Phylogenomic analyses reveal this lineage to belong to a widespread group of archaea that we propose to classify as a new euryarchaeal order ('Candidatus Altiarchaeales'). The representative, double-membraned species 'Candidatus Altiarchaeum hamiconexum' has an autotrophic metabolism that uses a not-yet-reported Factor420-free reductive acetyl-CoA pathway, confirmed by stable carbon isotopic measurements of archaeal lipids. Our results indicate that this lineage has evolved specific metabolic and structural features like nano-grappling hooks empowering this widely distributed archaeon to predominate anaerobic groundwater, where it may represent an important carbon dioxide sink.
Type I interferons have opposing effects during the emergence and recovery phases of colitis.
2014 - Eur J Immunol., 44: 2749-60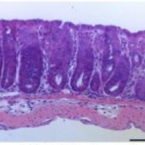
Abstract:
The contribution of the innate immune system to inflammatory bowel disease (IBD) is under intensive investigation. Research in animal models has demonstrated that type I interferons (IFN-Is) protect from IBD. In contrast, studies of patients with IBD have produced conflicting results concerning the therapeutic potential of IFN-Is. Here, we present data suggesting that IFN-Is play dual roles as regulators of intestinal inflammation in dextran sodium sulfate (DSS)-treated C57BL/6 mice. Though IFN-Is reduced acute intestinal damage and the abundance ofcolitis-associated intestinal bacteria caused by treatment with a high dose of DSS, they also inhibited the resolution of inflammation after DSS treatment. IFN-Is played an anti-inflammatory role by suppressing the release of IL-1β from the colon MHC class II(+) cells. Consistently, IL-1 receptor blockade reduced the severity of inflammation in IFN-I receptor-deficient mice and myeloid cell-restricted ablation of the IFN-I receptor was detrimental. The proinflammatory role of IFN-Is during recovery from DSS treatment was caused by IFN-I-dependent cell apoptosis as well as an increase in chemokine production and infiltrating inflammatory monocytes and neutrophils. Thus, IFN-Is play opposing roles in specificphases of intestinal injury and inflammation, which may be important for guiding treatment strategies in patients.
NanoSIMS combined with fluorescence microscopy as a tool for subcellular imaging of isotopically labeled platinum-based anticancer drugs
2014 - Chem. Sci., 5: 3135-3143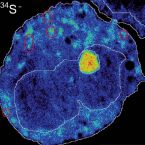
Abstract:
Multi-elemental, isotope selective nano-scale secondary ion mass spectrometry (NanoSIMS) combined with confocal laser-scanning microscopy was used to characterize the subcellular distribution of 15N-labeled cisplatin in human colon cancer cells. These analyses indicated predominant cisplatin colocalisation with sulfur-rich structures in both the nucleus and cytoplasm. Furthermore, colocalisation of platinum with phosphorus-rich chromatin regions was observed, which is consistent with its binding affinity to DNA as the generally accepted crucial target of the drug. Application of 15N-labeled cisplatin and subsequent measurement of the nitrogen isotopic composition and determination of the relative intensities of platinum and nitrogen associated secondary ion signals in different cellular compartments with NanoSIMS suggested partial dissociation of Pt–N bonds during the accumulation process, in particular within nucleoli at elevated cisplatin concentrations. This finding raises the question as to whether the observed intracellular dissociation of the drug has implications for the mechanism of action of cisplatin. Within the cytoplasm, platinum mainly accumulated in acidic organelles, as demonstrated by a direct combination of specific fluorescent staining, confocal laser scanning microscopy and NanoSIMS. Different processing of platinum drugs in acidic organelles might be relevant for their detoxification, as well as for their mode of action.
Growth of nitrite-oxidizing bacteria by aerobic hydrogen oxidation
2014 - Science, 345: 1052-1054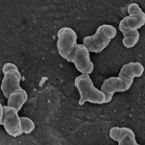
Abstract:
The bacterial oxidation of nitrite to nitrate is a key process of the biogeochemical nitrogen cycle. Nitrite-oxidizing bacteria are considered a highly specialized functional group, which depends on the supply of nitrite from other microorganisms and whose distribution strictly correlates with nitrification in the environment and in wastewater treatment plants. On the basis of genomics, physiological experiments, and single-cell analyses, we show that Nitrospira moscoviensis, which represents a widely distributed lineage of nitrite-oxidizing bacteria, has the genetic inventory to utilizehydrogen (H2) as an alternative energy source for aerobic respiration and grows on H2 without nitrite. CO2 fixation occurred with H2 as the sole electron donor. Our results demonstrate a chemolithoautotrophic lifestyle of nitrite-oxidizing bacteria outside the nitrogen cycle, suggesting greater ecological flexibility than previously assumed.
High-fat diet alters gut microbiota physiology in mice
2014 - ISME J., 8: 295-308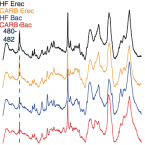
Abstract:
The intestinal microbiota is known to regulate host energy homeostasis and can be influenced by high-calorie diets. However, changes affecting the ecosystem at the functional level are still not well characterized. We measured shifts in cecal bacterial communities in mice fed a carbohydrate orhigh-fat (HF) diet for 12 weeks at the level of the following: (i) diversity and taxa distribution by high-throughput 16S ribosomal RNA gene sequencing; (ii) bulk and single-cell chemical composition by Fourier-transform infrared- (FT-IR) and Raman micro-spectroscopy and (iii) metaproteome and metabolome via high-resolution mass spectrometry. High-fat diet caused shifts in the diversity of dominant gut bacteria and altered the proportion of Ruminococcaceae (decrease) and Rikenellaceae (increase). FT-IR spectroscopy revealed that the impact of the diet on cecal chemical fingerprints is greater than the impact of microbiota composition. Diet-driven changes in biochemical fingerprints of members of the Bacteroidales and Lachnospiraceae were also observed at the level of single cells, indicating that there were distinct differences in cellular composition of dominant phylotypes under different diets. Metaproteome and metabolome analyses based on the occurrence of 1760 bacterial proteins and 86 annotated metabolites revealed distinct HF diet-specific profiles. Alteration of hormonal and anti-microbial networks, bile acid and bilirubin metabolism and shifts towards amino acid and simple sugars metabolism were observed. We conclude that a HF diet markedly affects the gut bacterial ecosystem at the functional level.
Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery
2014 - ISME J., 8(5):1101-14
Abstract:
Although alterations in gut microbiota composition during acute colitis have been repeatedly observed, associated functional changes and therecovery from dysbiosis received little attention. In this study, we investigated structure and function of the gut microbiota during acute inflammationand recovery in a dextran sodium sulfate (DSS)-colitis mouse model using metatranscriptomics, bacterial 16S rRNA gene amplicon sequencing and monitoring of selected host markers. Parallel to an increase of host markers of inflammation during acute colitis, we observed relative abundance shifts and alterations in phylotype composition of the dominant bacterial orders Clostridiales and Bacteroidales, and an increase of the low abundant Enterobacteriales, Deferribacterales, Verrucomicrobiales and Erysipelotrichales. During recovery, the microbiota began to resume, but did not reach its original composition until the end of the experiment. Microbial gene expression was more resilient to disturbance, with pre-perturbation-type transcript profiles appearing quickly after acute colitis. The decrease of Clostridiales during inflammation correlated with a reduction of transcripts related to butyrate formation, suggesting a disturbance in host-microbe signalling and mucosal nutrient provision. The impact of acute inflammationon the Clostridiales was also characterized by a significant downregulation of their flagellin-encoding genes. In contrast, the abundance of members of the Bacteroidales increased along with an increase in transcripts related to mucin degradation. We propose that acute inflammation triggered a selective reaction of the immune system against flagella of commensals and temporarily altered murine microbiota composition and functions relevant for the host. Despite changes in specific interactions, the host-microbiota homeostasis revealed a remarkable ability for recovery.
NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira
2014 - Environ Microbiol, 16: 3055-3071
Abstract:
Nitrospira are the most widespread and diverse known nitrite-oxidizing bacteria and key nitrifiers in natural and engineered ecosystems. Nevertheless, their ecophysiology and environmental distribution are understudied due to the recalcitrance of Nitrospira to cultivation and the lack of a molecular functional marker, which would allow the detection of Nitrospira in the environment. Here we introduce nxrB, the gene encoding subunit beta of nitrite oxidoreductase, as a functional and phylogenetic marker for Nitrospira. Phylogenetic trees based on nxrB of Nitrospira were largely congruent to 16S rRNA-based phylogenies. By using new nxrB-selective PCR primers, we obtained almost full-length nxrB sequences from Nitrospira cultures, two activated sludge samples, and several geographically and climatically distinct soils. Amplicon pyrosequencing of nxrB fragments from 16 soils revealed a previously unrecognized diversity of terrestrial Nitrospira with 1,801 detected species-level OTUs (using an inferred species threshold of 95% nxrB identity). Richness estimates ranged from 10 to 946 co-existing Nitrospira species per soil. Comparison to an archaeal amoA dataset obtained from the same soils [Environ. Microbiol. 14: 525-539 (2012)] uncovered that ammonia-oxidizing archaea and Nitrospira communities were highly correlated across the soil samples, possibly indicating shared habitat preferences or specific biological interactions among members of these nitrifier groups.
Enrichment and genome sequence of the group I.1a ammonia-oxidizing archaeon "Ca. Nitrosotenuis uzonensis" representing a clade globally distributed in thermal habitats
2013 - PLoS One, 8: e80835
Abstract:
The discovery of ammonia-oxidizing archaea (AOA) of the phylum Thaumarchaeota and the high abundance of archaeal ammonia monooxygenase subunit A encoding gene sequences in many environments have extended our perception of nitrifying microbial communities. Moreover, AOA are the only aerobic ammonia oxidizers known to be active in geothermal environments. Molecular data indicate that in many globally distributed terrestrial high-temperature habits a thaumarchaeotal lineage within the Nitrosopumilus cluster (also called marine group I.1a) thrives, but these microbes have neither been isolated from these systems nor functionally characterized in situ yet. In this study, we report on the enrichment and genomic characterization of a representative of this lineage from a thermal spring in Kamchatka. This thaumarchaeote, provisionally classified as "Candidatus Nitrosotenuis uzonensis", is a moderately thermophilic, non-halophilic, chemolithoautotrophic ammonia oxidizer. The nearly complete genome sequence (assembled into a single scaffold) of this AOA confirmed the presence of the typical thaumarchaeotal pathways for ammonia oxidation and carbon fixation, and indicated its ability to produce coenzyme F420 and to chemotactically react to its environment. Interestingly, like members of the genus Nitrosoarchaeum, "Candidatus N. uzonensis" also possesses a putative artubulin-encoding gene. Genome comparisons to related AOA with available genome sequences confirmed that the newly cultured AOA has an average nucleotide identity far below the species threshold and revealed a substantial degree of genomic plasticity with unique genomic regions in "Ca. N. uzonensis", which potentially include genetic determinants of ecological niche differentiation.
Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing
2013 - Proc. Natl. Acad. Sci. USA, 110: 4720-4725
Abstract:
The animal and human intestinal mucosa secretes an assortment of compounds to establish a physical barrier between the host tissue and intestinal contents, a separation that is vital for health. Some pathogenic microorganisms as well as members of the commensal intestinal microbiota have been shown to be able to break down these secreted compounds. Our understanding of host-compound degradation by the commensal microbiota has been limited to knowledge about simplified model systems because of the difficulty in studying the complex intestinal ecosystem in vivo. In this study, we introduce an approach that overcomes previous technical limitations and allows us to observe which microbial cells in the intestine use host-derived compounds. We added stable isotope-labeled threonine i.v. to mice and combined fluorescence in situ hybridization with high-resolution secondary ion mass spectrometry imaging to characterize utilization of host proteins by individual bacterial cells. We show that two bacterial species, Bacteroides acidifaciens and Akkermansia muciniphila, are important host-protein foragers in vivo. Using gnotobiotic mice we show that microbiota composition determines the magnitude and pattern of foraging by these organisms, demonstrating that a complex microbiota is necessary in order for this niche to be fully exploited. These results underscore the importance of in vivo studies of intestinal microbiota, and the approach presented in this study will be a powerful tool to address many other key questions in animal and human microbiome research.
Interactions of nitrifying bacteria and heterotrophs: Identification of a Micavibrio-like, putative predator of Nitrospira
2013 - Appl Environ Microbiol, 79: 2027-2037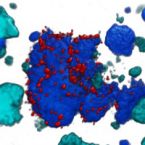
Abstract:
Chemolithoautotrophic nitrifying bacteria release soluble organic compounds, which can be substrates for heterotrophic microorganisms. The identity of these heterotrophs and the specificity of their interactions with nitrifiers are largely unknown. Here we incubated nitrifying activated sludge with (13)C-labeled bicarbonate and used stable isotope probing of 16S rRNA to follow the flow of carbon from uncultured nitrifiers to heterotrophs. To facilitate the identification of heterotrophs, the abundant 16S rRNA molecules from nitrifiers were depleted by catalytic oligonucleotides containing locked nucleic acids (LNAzymes), which specifically cut the 16S rRNA of defined target organisms. Among the (13)C-labeled heterotrophs were organisms remotely related to Micavibrio, a microbial predator of Gram-negative bacteria. Fluorescence in situ hybridization revealed a close spatial association of these organisms with microcolonies of nitrite-oxidizing sublineage I Nitrospira in sludge flocs. The high specificity of this interaction was confirmed by confocal microscopy and a novel image analysis method to quantify the localization patterns of biofilm microorganisms in 3-D space. Other isotope-labeled bacteria, which were affiliated with Thermomonas, co-localized less frequently with nitrifiers and thus were commensals or saprophytes rather than specific symbionts or predators. These results suggest that Nitrospira are subject to bacterial predation, which may influence the abundance and diversity of these nitrite oxidizers and the stability of nitrification in engineered and natural ecosystems. In silico screening of published next-generation sequencing datasets revealed a broad environmental distribution of the uncultured Micavibrio-like lineage.
Depletion of unwanted nucleic acid templates by selective cleavage: LNAzymes open a new window for detecting rare microbial community members
2013 - Appl. Environ. Microbiol., 79: 1534-1544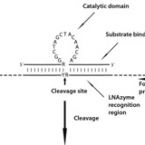
Abstract:
Many studies of molecular microbial ecology rely on the characterization of microbial communities by PCR amplification, cloning, sequencing, and phylogenetic analysis of genes encoding rRNAs or functional marker enzymes. However, if the established clone libraries are dominated by one or a few sequence types, the cloned diversity is difficult to analyze by random clone sequencing. Here we present a novel approach to deplete unwanted sequence types from complex nucleic acid mixtures prior to cloning and downstream analyses. It employs catalytically active oligonucleotides containing locked nucleic acids (LNAzymes) for the specific cleavage of selected RNA targets. When combined with in vitro transcription and reverse transcriptase PCR, this LNAzyme-based technique can be used with DNA or RNA extracts from microbial communities. The simultaneous application of more than one specific LNAzyme allows the concurrent depletion of different sequence types from the same nucleic acid preparation. This new method was evaluated with defined mixtures of cloned 16S rRNA genes and then used to identify accompanying bacteria in an enrichment culture dominated by the nitrite oxidizer "Candidatus Nitrospira defluvii." In silico analysis revealed that the majority of publicly deposited rRNA-targeted oligonucleotide probes may be used as specific LNAzymes with no or only minor sequence modifications. This efficient and cost-effective approach will greatly facilitate tasks such as the identification of microbial symbionts in nucleic acid preparations dominated by plastid or mitochondrial rRNA genes from eukaryotic hosts, the detection of contaminants in microbial cultures, and the analysis of rare organisms in microbial communities of highly uneven composition.
Zero-valent sulphur is a key intermediate in marine methane oxidation
2012 - Nature, 491: 541-546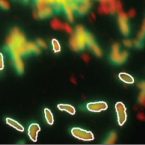
Abstract:
Emissions of methane, a potent greenhouse gas, from marine sediments are controlled by anaerobic oxidation of methane coupled primarily to sulphate reduction (AOM). Sulphate-coupled AOM is believed to be mediated by a consortium of methanotrophic archaea (ANME) and sulphate-reducing Deltaproteobacteria but the underlying mechanism has not yet been resolved. Here we show that zero-valent sulphur compounds (S0) are formed during AOM through a new pathway for dissimilatory sulphate reduction performed by the methanotrophic archaea. Hence, AOM might not be an obligate syntrophic process but may be carried out by the ANME alone. Furthermore, we show that the produced S0—in the form of disulphide—is disproportionated by the Deltaproteobacteria associated with the ANME. Our observations expand the diversity of known microbially mediated sulphur transformations and have significant implications for our understanding of the biogeochemical carbon and sulphur cycles.
Complete genome sequences of Desulfosporosinus orientis DSM765T, Desulfosporosinus youngiae DSM17734T, Desulfosporosinus meridiei DSM13257T, and Desulfosporosinus acidiphilus DSM22704T
2012 - J. Bacteriol., 194: 6300-1
Abstract:
Desulfosporosinus species are sulfate-reducing bacteria belonging to the Firmicutes. Their genomes will give insights into the genetic repertoire and evolution of sulfate reducers typically thriving in terrestrial environments and able to degrade toluene (Desulfosporosinus youngiae), to reduce Fe(III) (Desulfosporosinus meridiei, Desulfosporosinus orientis), and to grow under acidic conditions (Desulfosporosinus acidiphilus).
The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: Insights into metabolic versatility and environmental adaptations
2012 - Environ. Microbiol., 14: 3122-45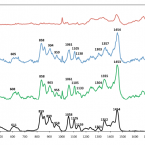
Abstract:
The cohort of the ammonia-oxidizing archaea (AOA) of the phylum Thaumarchaeota is a diverse, widespread and functionally important group of microorganisms in many ecosystems. However, our understanding of their biology is still very rudimentary in part because all available genome sequences of this phylum are from members of the Nitrosopumilus cluster. Here we report on the complete genome sequence of Candidatus Nitrososphaera gargensis obtained from an enrichment culture, representing a different evolutionary lineage of AOA frequently found in high numbers in many terrestrial environments. With its 2.83 Mb the genome is much larger than that of other AOA. The presence of a high number of (active) IS elements/transposases, genomic islands, gene duplications and a complete CRISPR/Cas defence system testifies to its dynamic evolution consistent with low degree of synteny with other thaumarchaeal genomes. As expected, the repertoire of conserved enzymes proposed to be required for archaeal ammonia oxidation is encoded by N. gargensis, but it can also use urea and possibly cyanate as alternative ammonia sources. Furthermore, its carbon metabolism is more flexible at the central pyruvate switch point, encompasses the ability to take up small organic compounds and might even include an oxidative pentose phosphate pathway. Furthermore, we show that thaumarchaeota produce cofactor F420 as well as polyhydroxyalkanoates. Lateral gene transfer from bacteria and euryarchaeota has contributed to the metabolic versatility of N. gargensis. This organisms is well adapted to its niche in a heavy metal-containing thermal spring by encoding a multitude of heavy metal resistance genes, chaperones and mannosylglycerate as compatible solute and has the genetic ability to respond to environmental changes by signal transduction via a large number of two-component systems, by chemotaxis and flagella-mediated motility and possibly even by gas vacuole formation. These findings extend our understanding of thaumarchaeal evolution and physiology and offer many testable hypotheses for future experimental research on these nitrifiers.
Modeling formamide denaturation of probe-target hybrids for improved microarray probe design in microbial diagnostics
2012 - PLoS One, 7: e43862
Abstract:
Application of high-density microarrays to the diagnostic analysis of microbial communities is challenged by the optimization of oligonucleotide probe sensitivity and specificity, as it is generally unfeasible to experimentally test thousands of probes. This study investigated the adjustment of hybridization stringency using formamide with the idea that sensitivity and specificity can be optimized during probe design if the hybridization efficiency of oligonucleotides with target and non-target molecules can be predicted as a function of formamide concentration. Sigmoidal denaturation profiles were obtained using fluorescently labeled and fragmented 16S rRNA gene amplicon of Escherichia coli as the target with increasing concentrations of formamide in the hybridization buffer. A linear free energy model (LFEM) was developed and microarray-specific nearest neighbor rules were derived. The model simulated formamide melting with a denaturant m-value that increased hybridization free energy (ΔG°) by 0.173 kcal/mol per percent of formamide added (v/v). Using the LFEM and specific probe sets, free energy rules were systematically established to predict the stability of single and double mismatches, including bulged and tandem mismatches. The absolute error in predicting the position of experimental denaturation profiles was less than 5% formamide for more than 90 percent of probes, enabling a practical level of accuracy in probe design. The potential of the modeling approach for probe design and optimization is demonstrated using a dataset including the 16S rRNA gene of Rhodobacter sphaeroides as an additional target molecule. The LFEM and thermodynamic databases were incorporated into a computational tool (ProbeMelt) that is freely available at http://DECIPHER.cee.wisc.edu
Nitrification expanded: Discovery, physiology, and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi
2012 - ISME J., 6: 2245-2256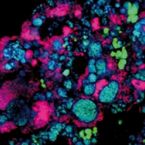
Abstract:
Nitrite-oxidizing bacteria (NOB) catalyze the second step of nitrification, a major process of the biogeochemical nitrogen cycle, but the recognized diversity of this guild is surprisingly low and only two bacterial phyla contain known NOB. Here, we report on the discovery of a chemolithoautotrophic nitrite oxidizer that belongs to the widespread phylum Chloroflexi not previously known to contain any nitrifying organism. This organism, named Nitrolancetus hollandicus, was isolated from a nitrifying reactor. Its tolerance to a broad temperature range (25-63 °C) and low affinity for nitrite (K(s)=1 mM), a complex layered cell envelope that stains Gram positive, and uncommon membrane lipids composed of 1,2-diols distinguish N. hollandicus from all other known nitrite oxidizers. N. hollandicus grows on nitrite and CO(2), and is able to use formate as a source of energy and carbon. Genome sequencing and analysis of N. hollandicus revealed the presence of all genes required for CO(2) fixation by the Calvin cycle and a nitrite oxidoreductase (NXR) similar to the NXR forms of the proteobacterial nitrite oxidizers, Nitrobacter and Nitrococcus. Comparative genomic analysis of the nxr loci unexpectedly indicated functionally important lateral gene transfer events between Nitrolancetus and other NOB carrying a cytoplasmic NXR, suggesting that horizontal transfer of the NXR module was a major driver for the spread of the capability to gain energy from nitrite oxidation during bacterial evolution. The surprising discovery of N. hollandicus significantly extends the known diversity of nitrifying organisms and likely will have implications for future research on nitrification in natural and engineered ecosystems.
Intracellular vesicles as reproduction elements in cell wall-deficient L-form bacteria
2012 - PLoS One, 7: e38514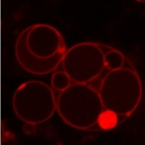
Abstract:
Cell wall-deficient bacteria, or L-forms, represent an extreme example of bacterial plasticity. Stable L-forms can multiply and propagate indefinitely in the absence of a cell wall. Data presented here are consistent with the model that intracellular vesicles in Listeria monocytogenes L-form cells represent the actual viable reproductive elements. First, small intracellular vesicles are formed along the mother cell cytoplasmic membrane, originating from local phospholipid accumulation. During growth, daughter vesicles incorporate a small volume of the cellular cytoplasm, and accumulate within volume-expanding mother cells. Confocal Raman microspectroscopy demonstrated the presence of nucleic acids and proteins in all intracellular vesicles, but only a fraction of which reveals metabolic activity. Following collapse of the mother cell and release of the daughter vesicles, they can establish their own membrane potential required for respiratory and metabolic processes. Premature depolarization of the surrounding membrane promotes activation of daughter cell metabolism prior to release. Based on genome resequencing of L-forms and comparison to the parental strain, we found no evidence for predisposing mutations that might be required for L-form transition. Further investigations revealed that propagation by intracellular budding not only occurs in Listeria species, but also in L-form cells generated from different Enterococcus species. From a more general viewpoint, this type of multiplication mechanism seems reminiscent of the physicochemical self-reproducing properties of abiotic lipid vesicles used to study the primordial reproduction pathways of putative prokaryotic precursor cells.
A straightforward DOPE-FISH method for simultaneous multicolor detection of six microbial populations
2012 - Appl. Environ. Microbiol., 78: 5138-42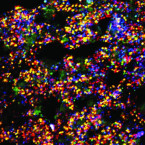
Abstract:
Fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes is an essential tool for the cultivation-independent identification of microbes within environmental and clinical samples. However, one of the major constraints of conventional FISH is the very limited number of different target organisms that can be detected simultaneously with standard epifluorescence or confocal laser scanning microscopy. Recently, this limitation has been overcome via an elegant approach termed combinatorial labeling and spectral imaging FISH (CLASI-FISH) (23). This technique, however, suffers compared to conventional FISH from an inherent loss in sensitivity and potential probe binding biases caused by the competition of two differentially labeled oligonucleotide probes for the same target site. Here we demonstrate that the application of multicolored, double-labeled oligonucleotide probes enables the simultaneous detection of up to six microbial target populations in a straightforward and robust manner with higher sensitivity and less bias. Thus, this newly developed technique should be an attractive option for all researchers interested in applying conventional FISH methods for the study of microbial communities.
Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis
2012 - ISME J., 6: 2091-106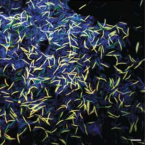
Abstract:
Human inflammatory bowel disease and experimental colitis models in mice are associated with shifts in intestinal microbiota composition, but it is unclear at what taxonomic/phylogenetic level such microbiota dynamics can be indicative for health or disease. Here, we report that dextran sodium sulfate (DSS)-induced colitis is accompanied by major shifts in the composition and function of the intestinal microbiota of STAT1(-/-) and wild-type mice, as determined by 454 pyrosequencing of bacterial 16S rRNA (gene) amplicons, metatranscriptomics and quantitative fluorescence in situ hybridization of selected phylotypes. The bacterial families Ruminococcaceae, Bacteroidaceae, Enterobacteriaceae, Deferribacteraceae and Verrucomicrobiaceae increased in relative abundance in DSS-treated mice. Comparative 16S rRNA sequence analysis at maximum possible phylogenetic resolution identified several indicator phylotypes for DSS treatment, including the putative mucin degraders Akkermansia and Mucispirillum. The analysis additionally revealed strongly contrasting abundance changes among phylotypes of the same family, particularly within the Lachnospiraceae. These extensive phylotype-level dynamics were hidden when reads were grouped at higher taxonomic levels. Metatranscriptomic analysis provided insights into functional shifts in the murine intestinal microbiota, with increased transcription of genes associated with regulation and cell signaling, carbohydrate metabolism and respiration and decreased transcription of flagellin genes during inflammation. These findings (i) establish the first in-depth inventory of the mouse gut microbiota and its metatranscriptome in the DSS colitis model, (ii) reveal that family-level microbial community analyses are insufficient to reveal important colitis-associated microbiota shifts and (iii) support a scenario of shifting intra-family structure and function in the phylotype-rich and phylogenetically diverse Lachnospiraceae in DSS-treated mice.
Sulfate-reducing microorganisms in wetlands - fameless actors in carbon cycling and climate change
2012 - Frontiers Microbiol., 3: 72
Abstract:
Freshwater wetlands are a major source of the greenhouse gas methane but at the same time can function as carbon sink. Their response to global warming and environmental pollution is one of the largest unknowns in the upcoming decades to centuries. In this review, we highlight the role of sulfate-reducing microorganisms (SRM) in the intertwined element cycles of wetlands. Although regarded primarily as methanogenic environments, biogeochemical studies have revealed a previously hidden sulfur cycle in wetlands that can sustain rapid renewal of the small standing pools of sulfate. Thus, dissimilatory sulfate reduction, which frequently occurs at rates comparable to marine surface sediments, can contribute up to 36–50% to anaerobic carbon mineralization in these ecosystems. Since sulfate reduction is thermodynamically favored relative to fermentative processes and methanogenesis, it effectively decreases gross methane production thereby mitigating the flux of methane to the atmosphere. However, very little is known about wetland SRM. Molecular analyses using dsrAB [encoding subunit A and B of the dissimilatory (bi)sulfite reductase] as marker genes demonstrated that members of novel phylogenetic lineages, which are unrelated to recognized SRM, dominate dsrAB richness and, if tested, are also abundant among the dsrAB-containing wetland microbiota. These discoveries point toward the existence of so far unknown SRM that are an important part of the autochthonous wetland microbiota. In addition to these numerically dominant microorganisms, a recent stable isotope probing study of SRM in a German peatland indicated that rare biosphere members might be highly active in situ and have a considerable stake in wetland sulfate reduction. The hidden sulfur cycle in wetlands and the fact that wetland SRM are not well represented by described SRM species explains their so far neglected role as important actors in carbon cycling and climate change.
amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions
2012 - Environ. Microbiol., 14: 525-539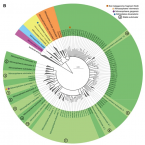
Abstract:
Ammonia-oxidizing archaea (AOA) play an important role in nitrification and many studies exploit their amoA genes as marker for their diversity and abundance. We present an archaeal amoA consensus phylogeny based on all publicly available sequences (status June 2010) and provide evidence for the diversification of AOA into four previously recognized clusters and one newly identified major cluster. These clusters, for which we suggest a new nomenclature, harbored 83 AOA species-level OTU (using an inferred species threshold of 85% amoA identity). 454 pyrosequencing of amoA amplicons from 16 soils sampled in Austria, Costa Rica, Greenland, and Namibia revealed that only 2% of retrieved sequences had no database representative on the species-level and represented 30–37 additional species-level OTUs. With the exception of an acidic soil from which mostly amoA amplicons of the Nitrosotalea cluster were retrieved, all soils were dominated by amoA amplicons from the Nitrososphaera cluster (also called group I.1b), indicating that the previously reported AOA from the Nitrosopumilus cluster (also called group I.1a) are absent or represent minor populations in soils. AOA richness estimates on the species level ranged from 8–83 co-existing AOAs per soil. Presence/absence of amoA OTUs (97% identity level) correlated with geographic location, indicating that besides contemporary environmental conditions also dispersal limitation across different continents and/or historical environmental conditions might influence AOA biogeography in soils.
New trends in fluorescence in situ hybridization for identification and functional analyses of microbes
2012 - Curr. Opinion. Biotech., 23: 96-102
Abstract:
Fluorescence in situ hybridization (FISH) has become an indispensable tool for rapid and direct single-cell identification of microbes by detecting signature regions in their rRNA molecules. Recent advances in this field include new web-based tools for assisting probe design and optimization of experimental conditions, easy-to-implement signal amplification strategies, innovative multiplexing approaches, and the combination of FISH with transmission electron microscopy or extracellular staining techniques. Further emerging developments focus on sorting FISH-identified cells for subsequent single-cell genomics and on the direct detection of specific genes within single microbial cells by advanced FISH techniques employing various strategies for massive signal amplification.
Bacteriocyte-associated gammaproteobacterial symbionts of the Adelges nordmannianae/piceae complex (Hemiptera: Adelgidae)
2012 - ISME J., 6: 384-396
Abstract:
Adelgids (Insecta: Hemiptera: Adelgidae) are known as severe pests of various conifers in North America, Canada, Europe and Asia. Here we present the first molecular identification of bacteriocyte-associated symbionts in these plant sap-sucking insects. Three geographically distant populations of members of the Adelges nordmannianae/piceae complex, identified based on coI and ef1alpha gene sequences, were investigated. Electron and light microscopy revealed two morphologically different endosymbionts, coccoid or polymorphic, that are located in distinct bacteriocytes. Phylogenetic analyses of their 16S and 23S rRNA gene sequences assigned both symbionts to novel lineages within the Gammaproteobacteria sharing less than 92% 16S rRNA sequence similarity with each other and showing no close relationship with known symbionts of insects. Their identity and intracellular location were confirmed by fluorescence in situ hybridisation, and the names ‘Candidatus Steffania adelgidicola’ and ‘Candidatus Ecksteinia adelgidicola’ are proposed for tentative classification. Both symbionts were present in all individuals of all investigated populations and in different adelgid life stages including eggs, suggesting vertical transmission from mother to offspring. An 85 kb genome fragment of ‘Candidatus Steffania adelgidicola’ was reconstructed based on a metagenomic library created from purified symbionts. Genomic features including the frequency of pseudogenes, the average length of intergenic regions and the presence of several genes, which are absent in other long-term obligate symbionts, suggested that ‘Candidatus Steffania adelgidicola’ is an evolutionary young bacteriocyte-associated symbiont, which has been acquired after the diversification of adelgids from their aphid sister group.
Chloroflexi bacteria are more diverse, abundant and similar in high than in low microbial abundance sponges
2011 - FEMS Microbiol. Ecol., 78: 497-510
Abstract:
Some marine sponges harbor dense and phylogenetically complex microbial communities [high microbial abundance (HMA) sponges] whereas others contain only few and less diverse microorganisms [low microbial abundance (LMA) sponges]. We focused on the phylum Chloroflexi that frequently occurs in sponges to investigate the different associations with three HMA and three LMA sponges from New Zealand. By applying a range of microscopical and molecular techniques a clear dichotomy between HMA and LMA sponges was observed: Chloroflexi bacteria were more abundant and diverse in HMA than in LMA sponges. Moreover, different HMA sponges contain similar Chloroflexi communities whereas LMA sponges harbor different and more variable communities which partly resemble Chloroflexi seawater communities. A comprehensive phylogenetic analysis of our own and publicly available sponge-derived Chloroflexi 16S rRNA gene sequences (> 780 sequences) revealed the enormous diversity of this phylum within sponges including 29 sponge-specific and sponge-coral clusters (SSC/SCC) as well as a 'supercluster' consisting of > 250 sponge-derived and a single nonsponge-derived 16S rRNA gene sequence. Interestingly, the majority of sequences obtained from HMA sponges, but only a few from LMA sponges, fell into SSC/SCC clusters. This indicates a much more specific association of Chloroflexi bacteria with HMA sponges and suggests an ecologically important role for these prominent bacteria.
Thaumarchaeotes abundant in refinery nitrifying sludges express amoA but are not obligate autotrophic ammonia oxidizers
2011 - Proc. Natl. Acad. Sci. USA, 108: 16771-16776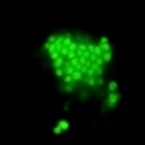
Abstract:
Nitrification is a core process in the global nitrogen cycle that is essential for the functioning of many ecosystems. The discovery of autotrophic ammonia-oxidizing archaea (AOA) within the phylum Thaumarchaeota has changed our perception of the microbiology of nitrification, in particular since their numerical dominance over ammonia-oxidizing bacteria (AOB) in many environments has been revealed. These and other data have led to a widely held assumption that all amoA-encoding members of the Thaumarchaeota (AEA) are autotrophic nitrifiers. In this study, 52 municipal and industrial wastewater treatment plants were screened for the presence of AEA and AOB. Thaumarchaeota carrying amoA were detected in high abundance only in four industrial plants. In one plant, thaumarchaeotes closely related to soil group I.1b outnumbered AOB up to 10,000-fold, and their numbers, which can only be explained by active growth in this continuous culture system, were two to three orders of magnitude higher than could be sustained by autotrophic ammonia oxidation. Consistently, (14)CO(2) fixation could only be detected in AOB but not in AEA in actively nitrifying sludge from this plant via FISH combined with microautoradiography. Furthermore, in situ transcription of archaeal amoA, and very weak in situ labeling of crenarchaeol after addition of (13)CO(2), was independent of the addition of ammonium. These data demonstrate that some amoA-carrying group I.1b Thaumarchaeota are not obligate chemolithoautotrophs.
Barcoded primers used in multiplex amplicon pyrosequencing bias amplification
2011 - Appl. Environ. Microbiol., 77: 7846-7849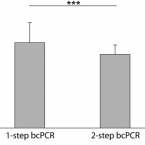
Abstract:
"Barcode-tagged" PCR primers used for multiplex amplicon sequencing generate a thus-far-overlooked amplification bias that produces variable terminal restriction fragment length polymorphism (T-RFLP) and pyrosequencing data from the same environmental DNA template. We propose a simple two-step PCR approach that increases reproducibility and consistently recovers higher genetic diversity in pyrosequencing libraries.
Systematic spatial bias in DNA microarray hybridization is caused by probe spot position-dependent variability in lateral diffusion
2011 - PLoS One, 6: e23727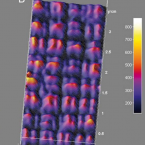
Abstract:
The hybridization of nucleic acid targets with surface-immobilized probes is a widely used assay for the parallel detection of multiple targets in medical and biological research. Despite its widespread application, DNA microarray technology still suffers from several biases and lack of reproducibility, stemming in part from an incomplete understanding of the processes governing surface hybridization. In particular, non-random spatial variations within individual microarray hybridizations are often observed, but the mechanisms underpinning this positional bias remain incompletely explained.
METHODOLOGY/PRINCIPAL FINDINGS:
This study identifies and rationalizes a systematic spatial bias in the intensity of surface hybridization, characterized by markedly increased signal intensity of spots located at the boundaries of the spotted areas of the microarray slide. Combining observations from a simplified single-probe block array format with predictions from a mathematical model, the mechanism responsible for this biasis found to be a position-dependent variation in lateral diffusion of target molecules. Numerical simulations reveal a strong influence of microarraywell geometry on the spatial bias.
CONCLUSIONS:
Reciprocal adjustment of the size of the microarray hybridization chamber to the area of surface-bound probes is a simple and effective measure to minimize or eliminate the diffusion-based bias, resulting in increased uniformity and accuracy of quantitative DNA microarrayhybridization.
Paracatenula, an ancient symbiosis between thiotrophic Alphaproteobacteria and catenulid flatworms
2011 - Proc. Natl. Acad. Sci. USA, 108: 12078-12083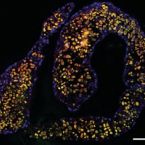
Abstract:
Harnessing chemosynthetic symbionts is a recurring evolutionary strategy. Eukaryotes from six phyla as well as one archaeon have acquired chemoautotrophic sulfur-oxidizing bacteria. In contrast to this broad host diversity, known bacterial partners apparently belong to two classes of bacteria-the Gamma- and Epsilonproteobacteria. Here, we characterize the intracellular endosymbionts of the mouthless catenulid flatworm genus Paracatenula as chemoautotrophic sulfur-oxidizing Alphaproteobacteria. The symbionts of Paracatenula galateia are provisionally classified as "Candidatus Riegeria galateiae" based on 16S ribosomal RNA sequencing confirmed by fluorescence in situ hybridization together with functional gene and sulfur metabolite evidence. 16S rRNA gene phylogenetic analysis shows that all 16 Paracatenula species examined harbor host species-specific intracellular Candidatus Riegeria bacteria that form a monophyletic group within the order Rhodospirillales. Comparing host and symbiont phylogenies reveals strict cocladogenesis and points to vertical transmission of the symbionts. Between 33% and 50% of the body volume of the various worm species is composed of bacterial symbionts, by far the highest proportion among all known endosymbiotic associations between bacteria and metazoans. This symbiosis, which likely originated more than 500 Mya during the early evolution of flatworms, is the oldest known animal-chemoautotrophic bacteria association. The distant phylogenetic position of the symbionts compared with other mutualistic or parasitic Alphaproteobacteria promises to illuminate the common genetic predispositions that have allowed several members of this class to successfully colonize eukaryote cells.
The Thaumarchaeota: An Emerging View of their Phylogeny and Ecophysiology
2011 - Curr. Opin. Microbiol., 14: 300-306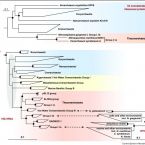
Abstract:
Thaumarchaeota range among the most abundant archaea on Earth. Initially classified as 'mesophilic Crenarchaeota', comparative genomics has recently revealed that they form a separate and deep-branching phylum within the Archaea. This novel phylum comprises in 16S rRNA gene trees not only all known archaeal ammonia oxidizers but also several clusters of environmental sequences representing microorganisms with unknown energy metabolism. Ecophysiological studies of ammonia-oxidizing Thaumarchaeota suggest adaptation to low ammonia concentrations and an autotrophic or possibly mixotrophic lifestyle. Extrapolating from the wide substrate range of copper-containing membrane-bound monooxygenases, to which the thaumarchaeal ammonia monooxygenases belong, the use of substrates other than ammonia for generating energy by some members of the Thaumarchaeota seems likely.
Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil
2011 - Proc. Natl. Acad. Sci. USA, 108: 8420-8425
Abstract:
Genes of archaea encoding homologues of ammonia monooxygenases have been found on a widespread basis and in large amounts in almost all terrestrial and marine environments, indicating that ammonia oxidizing archaea (AOA) might play a major role in nitrification on Earth. However, only one pure isolate of this group from a marine environment has so far been obtained, demonstrating archaeal ammonia oxidation coupled with autotrophic growth similar to the bacterial counterparts. Here we describe the cultivation and isolation of an AOA from soil. It grows on ammonia or urea as an energy source and is capable of using higher ammonia concentrations than the marine isolate, Nitrosopumilus maritimus. Surprisingly, although it is able to grow chemolithoautotrophically, considerable growth rates of this strain are obtained only upon addition of low amounts of pyruvate or when grown in coculture with bacteria. Our findings expand the recognized metabolic spectrum of AOA and help explain controversial results obtained in the past on the activity and carbon assimilation of these globally distributed organisms.
In situ techniques and digital image analysis methods for quantifying spatial localization patterns of nitrifiers and other microorganisms in biofilm and flocs
2011 - Methods Enzymol., 496: 185-215
Abstract:
The spatial localization patterns of microorganisms in multispecies biofilms reflect numerous phenomena that influence sessile microbial life, such as substrate concentration gradients within the biofilm and biological interactions with other biofilm populations. Quantitative and population-specific in situ analyses of spatial patterns have a high potential to provide novel insights into the biology of biofilm organisms, including yet uncultured microbes, but such approaches have been developed and used in a few studies only. Here, we outline digital image analysis methods to quantify the coaggregation, mutual avoidance, or random distribution of microbial populations in biofilm and flocs. A protocol is provided for fluorescence in situ hybridization with rRNA-targeted probes, which preserves the three-dimensional biofilm architecture for confocal microscopy and image analysis, and the combined use of these approaches is demonstrated by spatial analyses of nitrifying bacteria in complex biofilm samples.
Proteomic analysis reveals a virtually complete set of proteins for translation and energy generation in elementary bodies of the amoeba symbiont Protochlamydia amoebophila
2011 - Proteomics, 11: 1868-1892
Abstract:
Chlamydiae belong to the most successful intracellular bacterial pathogens. They display a complex developmental cycle and an extremely broad host spectrum ranging from vertebrates to protozoa. The family Chlamydiaceae comprises exclusively well-known pathogens of humans and animals, while members of its sister group, the Parachlamydiaceae, naturally occur as symbionts of free-living amoebae. Comparative analysis of these two groups provides valuable insights into chlamydial evolution and mechanisms for microbe-host interaction. Based on the complete genome sequence of the Acanthamoeba spp. symbiont Protochlamydia amoebophila UWE25, we performed the first detailed proteome analysis of the infectious stage of a symbiotic chlamydia. A 2-D reference proteome map was established and the analysis was extensively complemented by shotgun proteomics. In total, 472 proteins were identified, which represent 23.2% of all encoded proteins. These cover a wide range of functional categories, including typical house-keeping proteins, but also putative virulence-associated proteins. A number of proteins that are not encoded in genomes of Chlamydiaceae were observed and the expression of 162 proteins classified as hypothetical or unknown proteins could be demonstrated. Our findings indicate that P. amoebophila exploits its additional genetic repertoire (compared to the Chlamydiaceae), and that its elementary bodies are remarkably well equipped with proteins involved in transcription, translation, and energy generation.
Unexpected diversity of chlorite dismutases: A catalytically efficient dimeric enzyme from Nitrobacter winogradskyi
2011 - J. Bacteriol., 193: 2408-2417
Abstract:
Chlorite dismutase (Cld) is a unique heme enzyme catalyzing the conversion of ClO2- to Cl- and O2. Cld is usually found in perchlorate- or chlorate-reducing bacteria, but was recently identified also in a nitrite-oxidizing bacterium of the genus Nitrospira. Here we characterized a novel Cld-like protein from the chemolithoautotrophic nitrite oxidizer Nitrobacter winogradskyi, which is significantly smaller than all previously known chlorite dismutases. Its 3D crystal structure revealed a dimer of two identical subunits, which sharply contrasts the penta- or hexameric structures of other chlorite dismutases. Despite a truncated N-terminal domain in each subunit, this novel enzyme turned out to be a highly efficient chlorite dismutase (KM = 90 µM, kcat = 190 s-1, kcat/KM = 2.1 106 M-1 s-1), demonstrating a greater structural and phylogenetic diversity of these enzymes than previously known. Based on comparative analyses of Cld sequences and 3D structures, signature amino acid residues were identified that can be employed to assess whether uncharacterized Cld-like proteins may have a high chlorite-dismutating activity. Interestingly, proteins that contain all these signatures and are phylogenetically closely related to the novel-type Cld of N. winogradskyi exist in a large number of other microbes, including other nitrite oxidizers.
Microorganisms with novel dissimilatory (bi)sulfite reductase genes are widespread and part of the core microbiota in low-sulfate peatlands
2011 - Appl. Environ. Microbiol., 77: 1231-1242
Abstract:
Peatlands of the Lehstenbach catchment (Germany) house as-yet-unidentified microorganisms with phylogenetically novel variants of the dissimilatory (bi)sulfite reductase genes dsrAB. These genes are characteristic of microorganisms that reduce sulfate, sulfite, or some organosulfonates for energy conservation but can also be present in anaerobic syntrophs. However, nothing is currently known regarding the abundance, community dynamics, and biogeography of these dsrAB-carrying microorganisms in peatlands. To tackle these issues, soils from a Lehstenbach catchment site (Schlöppnerbrunnen II fen) from different depths were sampled at three time points over a 6-year period to analyze the diversity and distribution of dsrAB-containing microorganisms by a newly developed functional gene microarray and quantitative PCR assays. Members of novel, uncultivated dsrAB lineages (approximately representing species-level groups) (i) dominated a temporally stable but spatially structured dsrAB community and (ii) represented "core" members (up to 1% to 1.7% relative abundance) of the autochthonous microbial community in this fen. In addition, denaturing gradient gel electrophoresis (DGGE)- and clone library-based comparisons of the dsrAB diversity in soils from a wet meadow, three bogs, and five fens of various geographic locations (distance of 1 to 400 km) identified that one Syntrophobacter-related and nine novel dsrAB lineages are widespread in low-sulfate peatlands. Signatures of biogeography in dsrB-based DGGE data were not correlated with geographic distance but could be explained largely by soil pH and wetland type, implying that the distribution of dsrAB-carrying microorganisms in wetlands on the scale of a few hundred kilometers is not limited by dispersal but determined by local environmental conditions.
Proteomic analysis of the outer membrane of Protochlamydia amoebophila elementary bodies
2010 - Proteomics, 10: 4363-4376
Abstract:
Chlamydiae are obligate intracellular bacteria, comprising some of the most important bacterial pathogens of animals and humans. During their unique developmental cycle they have to attach to and enter their eukaryotic host cells, a process mediated by proteins in the chlamydial outer membrane. So far the only experimental data for chlamydial outer membrane proteins is available from members of the Chlamydiaceae, a family comprising exclusively human and animal pathogens. To get further insights into the evolution of the protein composition of the chlamydial outer membrane and into host-dependent differences, we performed an extensive experimental analysis of outer membrane fractions of Protochlamydia amoebophila elementary bodies, which constitute the infectious form of this non-pathogenic member of the Chlamydiae that thrives as a symbiont in Acanthamoeba spp. We used 1D and 2D gel electrophoresis in combination with MALDI-TOF, MALDI-TOF/TOF and nanoLC-ESI-MS/MS, and compared our experimental results with a previously published in silico analysis of chlamydial outer membrane proteins. This resulted in the identification of 38 proteins supported by both studies and therefore very likely to be located in the P. amoebophila outer membrane. The obtained experimental data provide the first comprehensive overview of outer membrane proteins of a chlamydial organism outside the Chlamydiaceae. They reveal both fundamental differences and convergent evolution between pathogenic and symbiotic chlamydiae.
Inclusion membrane proteins of Protochlamydia amoebophila UWE25 reveal a conserved mechanism for host cell interaction among the Chlamydiae
2010 - J. Bacteriol., 192: 5093-5102
Abstract:
Chlamydiae are a group of obligate intracellular bacteria comprising several important human pathogens. Inside the eukaryotic cell, chlamydiae remain within a host-derived vesicular compartment, termed the inclusion. They modify the inclusion membrane through insertion of unique proteins, which are involved in interaction with and manipulation of the host cell. Among chlamydiae, inclusion membrane proteins have been exclusively found in members of the family Chlamydiaceae, which predominantly infect mammalian and avian hosts. Here, the presence of inclusion membrane proteins in Protochlamydia amoebophila UWE25, a chlamydial endosymbiont of free-living amoebae, is reported. A genome-wide screening for secondary structure motifs resulted in the identification of 23 putative inclusion membrane proteins for this organism. Immunofluorescence analysis demonstrated that five of these proteins were expressed, and four of them could be localized to a halo surrounding the intracellular bacteria. Co-localisation studies showed an almost complete overlap of the signals obtained for the four putative inclusion membrane proteins, and immuno-transmission electron microscopy unambiguously demonstrated their location in the inclusion membrane. The presence of inclusion membrane proteins (designated IncA, IncQ, IncR, IncS) in P. amoebophila shows that this strategy for host cell interaction is conserved among the chlamydiae, and is used by chlamydial symbionts and pathogens alike.
A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria
2010 - Proc. Natl. Acad. Sci. USA, 107: 13479-13484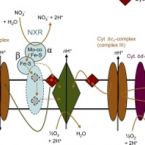
Abstract:
Nitrospira are barely studied and mostly uncultured nitrite-oxidizing bacteria, which are according to molecular data among the most diverse and widespread nitrifiers in natural ecosystems and biological wastewater treatment. Here, environmental genomics was used to reconstruct the complete genome of "Candidatus Nitrospira defluvii" from an activated sludge enrichment culture. Based on this first deciphered Nitrospira genome and on experimental data, we show that Ca. N. defluvii differs dramatically from other known nitrite oxidizers in the key enzyme nitrite oxidoreductase (NXR), the composition of the respiratory chain, and the pathway used for autotrophic carbon fixation, suggesting multiple independent evolution of chemolithoautotrophic nitrite oxidation. Adaptations of Ca. N. defluvii to substrate-limited conditions include a novel periplasmic NXR, which is constitutively expressed, and pathways for the transport, oxidation and assimilation of simple organic compounds that allow a mixotrophic lifestyle. The reverse tricarboxylic acid cycle as pathway for CO2 fixation and the lack of most classical defence mechanisms against oxidative stress suggest that Nitrospira evolved from microaerophilic or even anaerobic ancestors. Unexpectedly, comparative genomic analyses indicate functionally significant lateral gene transfer events between the genus Nitrospira and anaerobic ammonium-oxidizing planctomycetes, which share highly similar forms of NXR and other proteins reflecting that two key processes of the nitrogen cycle are evolutionary connected.
Structural and functional characterisation of the chlorite dismutase from the nitrite-oxidizing bacterium "Candidatus Nitrospira defluvii": Identification of a catalytically important amino acid residue
2010 - J. Struct. Biol., 172: 331-342
Abstract:
Chlorite dismutase (Cld) is a unique heme enzyme which transforms chlorite to chloride and molecular oxygen (reaction: ClO(2)(-)→Cl(-)+O(2)). Since bacteria with Cld play significant roles in the bioremediation of industrially contaminated sites and also in wastewater treatment, it is of high interest to understand the molecular mechanism of chlorite detoxification. Here we investigate a highly active Cld from Candidatus Nitrospira defluvii (NdCld), a key nitrifier in biological wastewater treatment, using a comprehensive structural, biochemical and bioinformatics approach. We determined the crystal structure of Cld from Candidatus Nitrospira defluvii and showed that functional NdCld is a homopentamer possessing a fold found in other Clds and Cld-like enzymes. To investigate the Cld function in more detail, site-directed mutagenesis of a catalytically important residue (Arg173) was performed and two enzyme mutants were structurally and biochemically characterized. Arginine 173 is demonstrated to play a key role in (i) controlling of ligand and substrate access and binding and (ii) in chlorite dismutation reaction. The flexible residue modulates the electrostatic potential and size of the active site entrance and might be involved in keeping transiently formed hypochlorite in place for final molecular oxygen and chloride formation. Furthermore, using a structure-based sequence alignment, we show that the residue corresponding to Arg173 is conserved in all known active forms of Cld and propose it as a marker for Cld activity in yet uncharacterized Cld-like proteins. Finally, our analysis indicates that all Clds and Cld-like enzymes employ a non-covalently bound heme as a cofactor.
Distinct gene set in two lineages of ammonia-oxidizing-archaea supports the phylum Thaumarchaeota
2010 - Trends Microbiol., 18: 331-340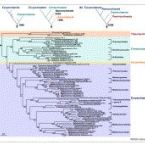
Abstract:
Globally distributed archaea comprising ammonia oxidizers of moderate terrestrial and marine environments are considered the most abundant archaeal organisms on Earth. Based on 16S rRNA phylogeny, initial assignment of these archaea was to the Crenarchaeota. By contrast, features of the first genome sequence from a member of this group suggested that they belong to a novel phylum, the Thaumarchaeota. Here, we re-investigate the Thaumarchaeota hypothesis by including two newly available genomes, that of the marine ammonia oxidizer Nitrosopumilus maritimus and that of Nitrososphaera gargensis, a representative of another evolutionary lineage within this group predominantly detected in terrestrial environments. Phylogenetic studies based on r-proteins and other core genes, as well as comparative genomics, confirm the assignment of these organisms to a separate phylum and reveal a Thaumarchaeota-specific set of core informational processing genes, as well as potentially ancestral features of the archaea.
Raman microspectroscopy reveals long-term extracellular activity of Chlamydiae.
2010 - Mol. Microbiol., 3: 687-700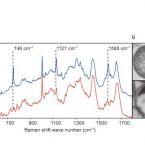
Abstract:
The phylum Chlamydiae consists exclusively of obligate intracellular bacteria. Some of them are formidable pathogens of humans, while others occur as symbionts of amoebae. These genetically intractable bacteria possess a developmental cycle consisting of replicative reticulate bodies and infectious elementary bodies, which are believed to be physiologically inactive. Confocal Raman microspectroscopy was applied to differentiate between reticulate bodies and elementary bodies of Protochlamydia amoebophila and to demonstrate in situ the labelling of this amoeba symbiont after addition of isotope-labelled phenylalanine. Unexpectedly, uptake of this amino acid was also observed for both developmental stages for up to 3 weeks, if incubated extracellularly with labelled phenylalanine, and P. amoebophila remained infective during this period. Furthermore, P. amoebophila energizes its membrane and performs protein synthesis outside of its host. Importantly, amino acid uptake and protein synthesis after extended extracellular incubation could also be demonstrated for the human pathogen Chlamydia trachomatis, which synthesizes stress-related proteins under these conditions as shown by 2-D gel electrophoresis and MALDI-TOF/TOF mass spectrometry. These findings change our perception of chlamydial biology and reveal that host-free analyses possess a previously not recognized potential for direct experimental access to these elusive microorganisms.
A 'rare biosphere' microorganism contributes to sulfate reduction in a peatland
2010 - ISME J., 12: 1591-1602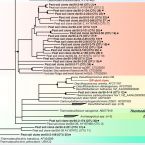
Abstract:
Methane emission from peatlands contributes substantially to global warming but is significantly reduced by sulfate reduction, which is fuelled by globally increasing aerial sulfur pollution. However, the biology behind sulfate reduction in terrestrial ecosystems is not well understood and the key players for this process as well as their abundance remained unidentified. Comparative 16S rRNA gene stable isotope probing (SIP) in the presence and absence of sulfate indicated that a Desulfosporosinus species, which constitutes only 0.006% of the total microbial community 16S rRNA genes, is an important sulfate reducer in a long-term experimental peatland field site. Parallel SIP using dsrAB (encoding subunit A and B of the dissimilatory (bi)sulfite reductase) identified no additional sulfate reducers under the conditions tested. For the identified Desulfosporosinus species a high cell-specific sulfate reduction rate of up to 341 fmol SO(4)(2-) cell(-1) day(-1) was estimated. Thus, the small Desulfosporosinus population has the potential to reduce sulfate in situ at a rate of 4.0-36.8 nmol (g soil w. wt.)(-1) day(-1), sufficient to account for a considerable part of sulfate reduction in the peat soil. Modeling of sulfate diffusion to such highly active cells identified no limitation in sulfate supply even at bulk concentrations as low as 10 muM. Collectively, these data show that the identified Desulfosporosinus species, despite being a member of the 'rare biosphere', contributes to an important biogeochemical process that diverts the carbon flow in peatlands from methane to CO(2) and, thus, alters their contribution to global warming.
Crenarchaeol dominates the membrane lipids of Candidatus Nitrososphaera gargensis, a thermophilic Group I.1b Archaeon
2010 - ISME J., 4: 542-552
Abstract:
Analyses of archaeal membrane lipids are increasingly being included in ecological studies as a comparatively unbiased complement to gene-based microbiological approaches. For example, crenarchaeol, a glycerol dialkyl glycerol tetraether (GDGT) with a unique cyclohexane moiety, has been postulated as biomarker for ammonia-oxidizing Archaea (AOA). Crenarchaeol has been detected in Nitrosopumilus maritimus and 'Candidatus Nitrosocaldus yellowstonii' representing two of the three lineages within the Crenarchaeota containing described AOA. In this paper we present the membrane GDGT composition of 'Candidatus Nitrososphaera gargensis', a moderately thermophilic AOA, and the only cultivated Group I.1b Crenarchaeon. At a cultivation temperature of 46 degrees C, GDGTs of this organism consisted primarily of crenarchaeol, its regioisomer, and a novel GDGT. Intriguingly, 'Ca. N. gargensis' is the first cultivated archaeon to synthesize substantial amounts of the crenarchaeol regioisomer, a compound found in large relative abundances in tropical ocean water and some soils, and an important component of the TEX(86) paleothermometer. Intact polar lipid (IPL) analysis revealed that 'Ca. N. gargensis' synthesizes IPLs similar to those reported for the Goup I.1a AOA, Nitrosopumilus maritimus SCMI, in addition to IPLs containing uncharacterized headgroups. Overall, the unique GDGT composition of 'Ca. N. gargensis' extends the known taxonomic distribution of crenarchaeol synthesis to the Group I.1b Crenarchaeota, implicating this clade as a potentially important source of crenarchaeol in soils and moderately high temperature environments. Moreover, this work supports the hypothesis that crenarchaeol is specific to all AOA and highlights specific lipids, which may prove useful as biomarkers for 'Ca. N. gargensis'-like AOA.
The genome of the amoeba symbiont 'Candidatus Amoebophilus asiaticus' reveals common mechanisms for host cell interaction among amoeba-associated bacteria
2010 - J. Bacteriol., 192: 1045-1057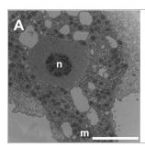
Abstract:
Protozoa play host for many intracellular bacteria and are important for the adaptation of pathogenic bacteria to eukaryotic cells. We analyzed the genome sequence of "Candidatus Amoebophilus asiaticus," an obligate intracellular amoeba symbiont belonging to the Bacteroidetes. The genome has a size of 1.89 Mbp, encodes 1,557 proteins, and shows massive proliferation of IS elements (24% of all genes), although the genome seems to be evolutionarily relatively stable. The genome does not encode pathways for de novo biosynthesis of cofactors, nucleotides, and almost all amino acids. "Ca. Amoebophilus asiaticus" encodes a variety of proteins with predicted importance for host cell interaction; in particular, an arsenal of proteins with eukaryotic domains, including ankyrin-, TPR/SEL1-, and leucine-rich repeats, which is hitherto unmatched among prokaryotes, is remarkable. Unexpectedly, 26 proteins that can interfere with the host ubiquitin system were identified in the genome. These proteins include F- and U-box domain proteins and two ubiquitin-specific proteases of the CA clan C19 family, representing the first prokaryotic members of this protein family. Consequently, interference with the host ubiquitin system is an important host cell interaction mechanism of "Ca. Amoebophilus asiaticus". More generally, we show that the eukaryotic domains identified in "Ca. Amoebophilus asiaticus" are also significantly enriched in the genomes of other amoeba-associated bacteria (including chlamydiae, Legionella pneumophila, Rickettsia bellii, Francisella tularensis, and Mycobacterium avium). This indicates that phylogenetically and ecologically diverse bacteria which thrive inside amoebae exploit common mechanisms for interaction with their hosts, and it provides further evidence for the role of amoebae as training grounds for bacterial pathogens of humans.
Double-labeling of oligonucleotide probes for fluorescence in situ hybridization (DOPE-FISH) improves signal intensity and increases rRNA accessibility
2010 - Appl. Environ. Microbiol., 76: 922–926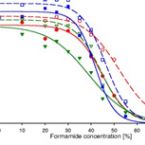
Abstract:
Fluorescence in situ hybridization (FISH) with singly labeled rRNA-targeted oligonucleotide probes is widely applied for direct identification of microbes in the environment or in clinical specimens. Here we show that a replacement of singly labeled oligonucleotide probes with 5'-, 3'-doubly labeled probes at least doubles FISH signal intensity without causing specificity problems. Furthermore, Cy3-doubly labeled probes strongly increase in situ accessibility of rRNA target sites and thus provide more flexibility for probe design.
Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts
2010 - Environ. Microbiol., 12: 2070-2082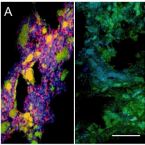
Abstract:
Marine sponges contain complex bacterial communities of considerable ecological and biotechnological importance, with many of these organisms postulated to be specific to sponge hosts. Testing this hypothesis in light of the recent discovery of the rare microbial biosphere, we investigated three Australian sponges by massively parallel 16S rRNA gene tag pyrosequencing. Here we show bacterial diversity that is unparalleled in an invertebrate host, with more than 250,000 sponge-derived sequence tags being assigned to 23 bacterial phyla and revealing up to 2996 operational taxonomic units (95% sequence similarity) per sponge species. Of the 33 previously described sponge-specific clusters that were detected in this study, 48% were found exclusively in adults and larvae implying vertical transmission of these groups. The remaining taxa, including Poribacteria, were also found at very low abundance among the 135,000 tags retrieved from surrounding seawater. Thus, members of the rare seawater biosphere may serve as seed organisms for widely occurring symbiont populations in sponges and their host association might have evolved much more recently than previously thought.
Single-cell ecophysiology of microbes as revealed by Raman microspectroscopy or secondary ion mass spectrometry imaging
2009 - Annu Rev Microbiol., 63: 411-29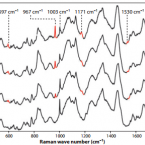
Abstract:
An astonishing diversity of microorganisms thrives on our planet and their activities are fundamental for the functioning of all ecosystems including the human body. Consequently, detailed insights into the functions performed by microorganisms in their natural environment are required to understand human biology and the biology of the world around us and to lay the foundations for targeted manipulation of microbial communities. Isotope-labeling techniques combined with molecular detection tools are frequently used by microbial ecologists to directly link structure and function of microbial communities and to monitor metabolic properties of uncultured microbes at the single-cell level. However, only the recent combination of such techniques with Raman microspectroscopy or secondary ion mass spectrometry enables functional studies of microbes on a single-cell level by using stable isotopes as labels. This review provides an overview of these new techniques and their applications in microbial ecology, which allow us to investigate the ecophysiology of uncultured microbes to an extent that was unimaginable just a few years ago.
Comprehensive in silico prediction and analysis of chlamydial outer membrane proteins reflects evolution and life style of the Chlamydiae
2009 - BMC Genomics, 10: 634
Abstract:
Chlamydiae are obligate intracellular bacteria comprising some of the most important bacterial pathogens of animals and humans. Although chlamydial outer membrane proteins play a key role for attachment to and entry into host cells, only few have been described so far. We developed a comprehensive, multiphasic in silico approach, including the calculation of clusters of orthologues, to predict outer membrane proteins using conservative criteria. We tested this approach using Escherichia coli (positive control) and Bacillus subtilis (negative control), and applied it to five chlamydial species; Chlamydia trachomatis, Chlamydia muridarum, Chlamydia (a.k.a. Chlamydophila) pneumoniae, Chlamydia (a.k.a. Chlamydophila) caviae, and Protochlamydia amoebophila. In total, 312 chlamydial outer membrane proteins and lipoproteins in 88 orthologous clusters were identified, including 238 proteins not previously recognized to be located in the outer membrane. Analysis of their taxonomic distribution revealed an evolutionary conservation among Chlamydiae, Verrucomicrobia, Lentisphaerae and Planctomycetes as well as lifestyle-dependent conservation of the chlamydial outer membrane protein composition. This analysis suggested a correlation between the outer membrane protein composition and the host range of chlamydiae and revealed a common set of outer membrane proteins shared by these intracellular bacteria. The collection of predicted chlamydial outer membrane proteins is available at the online database pCOMP (www.microbial-ecology.net/pcomp) and might provide future guidance in the quest for anti-chlamydial vaccines.
Isotope array analysis of Rhodocyclales uncovers functional redundancy and versatility in an activated sludge
2009 - ISME J., 3: 1349-1364
Abstract:
Extensive physiological analyses of different microbial community members in many samples are difficult because of the restricted number of target populations that can be investigated in reasonable time by standard substrate-mediated isotope-labeling techniques. The diversity and ecophysiology of Rhodocyclales in activated sludge from a full-scale wastewater treatment plant were analyzed following a holistic strategy based on the isotope array approach, which allows for a parallel functional probing of different phylogenetic groups. Initial diagnostic microarray, comparative 16S rRNA gene sequence, and quantitative fluorescence in situ hybridization surveys indicated the presence of a diverse community, consisting of an estimated number of 27 operational taxonomic units that grouped in at least seven main Rhodocyclales lineages. Substrate utilization profiles of probe-defined populations were determined by radioactive isotope array analysis and microautoradiography-fluorescence in situ hybridization of activated sludge samples that were briefly exposed to different substrates under oxic and anoxic, nitrate-reducing conditions. Most detected Rhodocyclales groups were actively involved in nitrogen transformation, but varied in their consumption of propionate, butyrate, or toluene, and thus in their ability to use different carbon sources in activated sludge. This indicates that the functional redundancy of nitrate reduction and the functional versatility of substrate usage are important factors governing niche overlap and differentiation of diverse Rhodocyclales members in this activated sludge.
High genetic similarity between two geographically distinct strains of the sulfur-oxidizing symbiont 'Candidatus Thiobios zoothamnicoli'
2009 - FEMS Microbiol. Ecol., 67: 229-41
Abstract:
The giant marine ciliate Zoothamnium niveum (Ciliophora, Oligohymenophora) is obligatorily covered by a monolayer of putative chemoautotrophicsulfur-oxidizing (thiotrophic) bacteria. For Z. niveum specimens from the Caribbean Sea it has been demonstrated that this ectosymbiotic population consists of only a single pleomorphic phylotype described as Candidatus Thiobios zoothamnicoli. The goal of our study was to identify and phylogenetically analyse the ectosymbiont(s) of a recently discovered Z. niveum population from the Mediterranean Sea, and to compare marker genes encoding key enzymes of the carbon and sulfur metabolism between the two symbiont populations. We identified a single bacterial phylotype representing the ectosymbiont of Z. niveum from the Mediterranean population showing 99.7% 16S rRNA gene (99.2% intergenic spacer region)similarity to the Caribbean Z. niveum ectosymbiont. Genes encoding enzymes typical for an inorganic carbon metabolism [ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO)] and for sulfur metabolism (5'-adenylylsulfate reductase, dissimilatory sulfite reductase) were detected in both symbiotic populations. The very high amino acid sequence identity (97-100%) and the high nucleic acid sequence identity (90-98%) of these marker enzymes in two geographically distant symbiont populations suggests that the association of Z. niveum with Cand. Thiobios zoothamnicoli is very specific as well as temporally and spatially stable.
Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes
2009 - Environ. Microbiol., 11: 289-299
Abstract:
Sulfur-oxidizing prokaryotes (SOP) catalyse a central step in the global S-cycle and are of major functional importance for a variety of natural and engineered systems, but our knowledge on their actual diversity and environmental distribution patterns is still rather limited. In this study we developed a specific PCR assay for the detection of dsrAB that encode the reversely operating sirohaem dissimilatory sulfite reductase (rDSR) and are present in many but not all published genomes of SOP. The PCR assay was used to screen 42 strains of SOP (most without published genome sequence) representing the recognized diversity of this guild. For 13 of these strains dsrAB was detected and the respective PCR product was sequenced. Interestingly, most dsrAB-encoding SOP are capable of forming sulfur storage compounds. Phylogenetic analysis demonstrated largely congruent rDSR and 16S rRNA consensus tree topologies, indicating that lateral transfer events did not play an important role in the evolutionary history of known rDSR. Thus, this enzyme represents a suitable phylogenetic marker for diversity analyses of sulfur storage compound-exploiting SOP in the environment. The potential of this new functional gene approach was demonstrated by comparative sequence analyses of all dsrAB present in published metagenomes and by applying it for a SOP census in selected marine worms and an alkaline lake sediment.
probeCheck - a central resource for evaluating oligonucleotide probe coverage and specificity
2008 - Environ. Microbiol., 10: 2894-2896
Abstract:
The web server probeCheck, freely accessible at www.microbial-ecology.net/probecheck, provides a pivotal forum for rapid specificity and coverage evaluations of probes and primers against selected databases of phylogenetic and functional marker genes. Currently, 24 widely used sequence collections including the Ribosomal Database Project (RDP) II, Greengenes, SILVA, and the Functional Gene Pipeline/Repository can be queried. For this purpose, probeCheck integrates a new online version of the popular ARB probe match tool with free energy (ΔG) calculations for each perfectly-matched and mismatched probe-target hybrid, allowing assessment of the theoretical binding stabilities of oligo-target and non-target hybrids. For each output sequence, the accession number, the GenBank taxonomy, and a link to the respective entry at GenBank, EMBL, and, if applicable, the query database is displayed. Filtering options allow customizing results on the output page. In addition, probeCheck is linked with probe match tools of RDP II and Greengenes, NCBI Blast, the Oligonucleotide Properties Calculator, the two-state folding tool of the DINAMelt server, and the rRNA-targeted probe database probeBase. Taken together, these features provide a multifunctional platform with maximal flexibility for the user in the choice of databases and options for the evaluation of published and newly developed probes and primers.
Diversity of bacterial endosymbionts of environmental Acanthamoeba isolates
2008 - Appl. Environ. Microbiol., 74: 5822-5831
Abstract:
Free-living amoebae are frequent hosts for bacterial endosymbionts. In this study, the symbionts of eight novel environmental Acanthamoeba strains isolated from different locations worldwide were characterized. Phylogenetic analysis revealed that they were related to either of four evolutionary lineages of amoeba symbionts recognized previously. This study provides evidence for the existence of only a small number of phylogenetically well separated groups of obligate intracellular endosymbionts of acanthamoebae with global distribution.
Lawsonia intracellularis encodes a functional rickettsia-like ATP/ADP translocase for host exploitation
2008 - J. Bacteriol., 190: 5746-5752
Abstract:
ATP/ADP translocases are a hallmark of obligate intracellular pathogens related to chlamydiae and rickettsiae. These proteins catalyze the highly specific exchange of bacterial ADP against host ATP and thus allow bacteria to exploit the hosts’ energy pool, a process also referred to as energy parasitism. The genome sequence of the obligate intracellular pathogen Lawsonia intracellularis (Deltaproteobacteria), responsible for one of the most economically important diseases in swine industry worldwide, revealed the presence of a putative ATP/ADP translocase most similar to known ATP/ADP translocases of chlamydiae and rickettsiae (around 47% amino acid sequence identity). The gene coding for the putative ATP/ADP translocase of L. intracellularis (LiNTT1) was cloned and expressed in the heterologous host Escherichia coli. Transport properties of LiNTT1 were determined by measuring uptake of radioactively labeled substrates into E. coli. LiNTT1 transported ATP in a counter exchange mode with ADP in a highly specific manner; determined substrate affinities were 236.3 (± 36.5) µM for ATP and 275.2 (± 28.1) µM for ADP, identifying this protein as functional ATP/ADP translocase. LiNTT1 is the first ATP/ADP translocase from a bacterium not related to Chlamydiae or Rickettsiales, showing that energy parasitism by ATP/ADP translocases is more widespread than previously recognized. The occurrence of an ATP/ADP translocase in L. intracellularis is explained by a relatively recent horizontal gene transfer event, with rickettsiae as donor.
Quantification of target molecules needed to detect microorganisms by fluorescence in situ hybridization (FISH) and catalyzed reporter deposition-FISH
2008 - Appl. Environ. Microbiol., 74: 5068-5077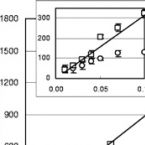
Abstract:
Fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes is a widely used method to detect and quantify microorganisms by fluorescence microscopy in environmental samples and medical specimens. Difficulties with FISH arise if the rRNA content of the probe-target organisms is low, causing dim fluorescence signals that are not detectable against background fluorescence. This limitation is ameliorated by technical modifications such as catalysed reporter deposition-fluorescence in situ hybridization (CARD-FISH), but to date the minimal rRNA copy numbers needed to obtain a visible signal of a microbial cell after FISH or CARD-FISH have not been determined. In this study, a novel competitive FISH approach was developed and used to quantify, based on a thermodynamic model of probe competition, the 16S rRNA copy numbers per cell required to detect bacteria by FISH and CARD-FISH with oligonucleotide probes in mixed pure cultures and in activated sludge. The detection limit of conventional FISH with Cy3-labelled probe EUB338-I was found to be 370±45 16S rRNA molecules per cell for E. coli hybridized on microscope glass slides, and 1,400±170 16S rRNA copies per E. coli cell in activated sludge. For CARD-FISH the respective values ranged from 8.9±1.5 to 14±2 and from 36±6 to 54±7 16S rRNA molecules per cell, indicating that the sensitivity of CARD-FISH was 26 to 41-fold higher than that of conventional FISH. These results suggest that optimized FISH protocols using oligonucleotide probes could be suitable for more recent applications of FISH, for example to detect mRNA in situ in microbial cells.
Microbial diversity and the genetic nature of microbial species
2008 - Nature Rev. Microbiol., 6: 431-440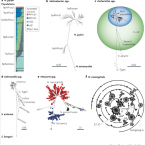
Abstract:
The earth contains a huge number of largely uncharacterized Bacteria and Archaea. Microbiologists are struggling to summarize their genetic diversity and classify them, which has resulted in heated debates on methods for defining species, mechanisms that lead to speciation and whether microbial species even exist. This Review proposes that decisions on the existence of species and methods to define them should be guided by a method-free species concept that is based on cohesive evolutionary forces. It summarizes current approaches to defining species and the problems of these approaches, and presents selected examples of the population genetic patterns at and below the species level.
Environmental genomics reveals a functional chlorite dismutase in the nitrite-oxidizing bacterium "Candidatus Nitrospira defluvii"
2008 - Environ. Microbiol., 10: 3043-3056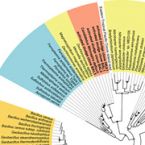
Abstract:
Nitrite-oxidizing bacteria of the genus Nitrospira are ubiquitous in natural ecosystems and also in wastewater treatment plants. Nitrospira are members of a distinct phylum, not closely related to other nitrifiers, and no genomic sequences from this genus have been available so far. Here we applied an environmental genomics approach to sequence and assemble a 137 kbp-long genome fragment of 'Candidatus Nitrospira defluvii', which had been enriched from activated sludge and belongs to Nitrospira sublineage I without isolated representatives. The annotation of this contig, which carried the 16S rRNA gene of N. defluvii, offered first insight into the genome of Nitrospira. Surprisingly, we found a gene similar to genes encoding chlorite dismutase (CLD), an enzyme degrading chlorite (ClO(2)(-)) to Cl(-) and O(2). To date, CLDs with high catalytic activity have been found only in perchlorate- and chlorate-reducing bacteria but not in nitrifiers. Heterologous expression in E. coli followed by enzymatic tests confirmed that this gene of Nitrospira encodes a highly active CLD, which is also expressed in situ by Nitrospira, indicating that this nitrite oxidizer might be involved in the bioremediation of perchlorate and chlorite. Phylogenetic analyses showed that CLD and related proteins are widely distributed among the Bacteria and Archaea, and indicated that this enzyme family appeared relatively early in evolution, has been subject to functional diversification and might play yet unknown roles in microbial metabolism.
Chlamydia-like bacteria in respiratory samples of community-acquired pneumonia patients
2008 - FEMS Microbiol. Lett., 281: 198-202
Abstract:
Chlamydia-like bacteria, obligate intracellular relatives of Chlamydia trachomatis and Chlamydophila pneumoniae, are widely distributed in nature. Using a two-step nested and semi-nested PCR approach targeting the 16S rRNA gene, we found DNA of chlamydia-like bacteria in respiratory samples from patients with community-acquired pneumonia. Four out of 387 cases (1.03%) tested positive if only sequences showing less than 99.9% 16S rRNA sequence similarity to known chlamydiae were considered. These included for the first time Protochlamydia amoebophila, Waddlia chondrophila, and ‘Candidatus Rhabdochlamydia porcellionis’-related sequences. This study extends previous findings suggesting an association of chlamydia-like bacteria with respiratory disease, but a causal link between these microorganisms and respiratory tract infections has yet to be established.
A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring
2008 - Proc. Natl. Acad. Sci. USA, 105: 2134-2139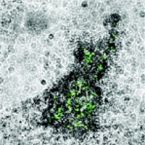
Abstract:
The recent discovery of ammonia-oxidizing archaea (AOA) dramatically changed our perception of the diversity and evolutionary history of microbes involved in nitrification. In this study, a moderately thermophilic (46°C) ammonia-oxidizing enrichment culture, which had been seeded with biomass from a hot spring, was screened for ammonia oxidizers. Although gene sequences for crenarchaeotal 16S rRNA and two subunits of the ammonia monooxygenase (amoA and amoB) were detected via PCR, no hints for known ammonia-oxidizing bacteria were obtained. Comparative sequence analyses of these gene fragments demonstrated the presence of a single operational taxonomic unit and thus enabled the assignment of the amoA and amoB sequences to the respective 16S rRNA phylotype, which belongs to the widely distributed group I.1b (soil group) of the Crenarchaeota. Catalyzed reporter deposition (CARD)–FISH combined with microautoradiography (MAR) demonstrated metabolic activity of this archaeon in the presence of ammonium. This finding was corroborated by the detection of amoA gene transcripts in the enrichment. CARD-FISH/MAR showed that the moderately thermophilic AOA is highly active at 0.14 and 0.79 mM ammonium and is partially inhibited by a concentration of 3.08 mM. The enriched AOA, which is provisionally classified as "Candidatus Nitrososphaera gargensis," is the first described thermophilic ammonia oxidizer and the first member of the crenarchaeotal group I.1b for which ammonium oxidation has been verified on a cellular level. Its preference for thermophilic conditions reinvigorates the debate on the thermophilic ancestry of AOA.
Diversity and mode of transmission of ammonia-oxidizing archaea in marine sponges
2008 - Environ. Microbiol., 10: 1087-1094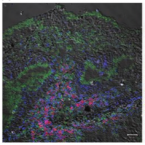
Abstract:
The model marine crenarchaeote 'Cenarchaeum symbiosum' is until now the only ammonia-oxidizing archaeon known from a marine sponge. Here, phylogenetic analyses based on the 16S rRNA and ammonia monooxygenase subunit A (amoA) genes revealed the presence of putative ammonia-oxidizing archaea (AOA) in a diverse range of sponges from the western Pacific, Caribbean and Mediterranean. amoA diversity was limited even between different oceans, with many of the obtained sequences (75.9%; n(total) = 83) forming a monophyletic, apparently sponge- (and coral-) specific lineage, analogous to those previously inferred from comparative 16S rRNA gene studies of sponge-associated microbes. The presence of AOA in sponge larvae, as detected by 16S rRNA and amoA PCR assays as well as by fluorescence in situ hybridization, suggests they are vertically transmitted and thus might be of importance for ammonia detoxification within the sponge.
Raman-FISH: combining stable-isotope Raman spectroscopy and fluorescence in situ hybridization for the single cell analysis of identity and function
2007 - Environ Microbiol., 9: 1878-1889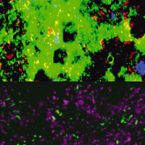
Abstract:
We have coupled fluorescence in situ hybridization (FISH) with Raman microscopy for simultaneous cultivation-independent identification and determination of (13)C incorporation into microbial cells. Highly resolved Raman confocal spectra were generated for individual cells which were grown in minimal medium where the ratio of (13)C to (12)C content of the sole carbon source was incrementally varied. Cells which were (13)C-labelled through anabolic incorporation of the isotope exhibited key red-shifted spectral peaks, the calculated 'red shift ratio' (RSR) being highly correlated with the (13)C-content of the cells. Subsequently, Raman instrumentation and FISH protocols were optimized to allow combined epifluorescence and Raman imaging of Fluos, Cy3 and Cy5-labelled microbial populations at the single cell level. Cellular (13)C-content determinations exhibited good congruence between fresh cells and FISH hybridized cells indicating that spectral peaks, including phenylalanine resonance, which were used to determine (13)C-labelling, were preserved during fixation and hybridization. In order to demonstrate the suitability of this technology for structure-function analyses in complex microbial communities, Raman-FISH was deployed to show the importance of Pseudomonas populations during naphthalene degradation in groundwater microcosms. Raman-FISH extends and complements current technologies such as FISH-microautoradiography and stable isotope probing in that it can be applied at the resolution of single cells in complex communities, is quantitative if suitable calibrations are performed, can be used with stable isotopes and has analysis times of typically 1 min per cell.
Who eats what, where and when? Isotope-labelling experiments are coming of age
2007 - ISME J., 1: 103-110
Abstract:
Isotope-labelling experiments have changed the way microbial ecologists investigate the ecophysiology of microbial populations and cells in the environment. Insight into the 'uncultivated majority' accompanies methodology that involves the incorporation of stable isotopes or radioisotopes into sub-populations of environmental samples. Subsequent analysis of labelled biomarkers of sub-populations with stable-isotope probing (DNA-SIP, RNA-SIP, phospholipid-derived fatty acid-SIP) or individual cells with a combination of fluorescence in situ hybridization and microautoradiography reveals linked phylogenetic and functional information about the organisms that assimilated these compounds. Here, we review some of the most recent literature, with an emphasis on methodological improvements to the sensitivity and utility of these methods. We also highlight related isotope techniques that are in continued development and hold promise to transform the way we link phylogeny and function in complex microbial communities.
Sponge-associated microorganisms: evolution, ecology and biotechnological potential
2007 - Microbiol. Mol. Biol. Rev., 71: 295-347
Abstract:
Marine sponges often contain diverse and abundant microbial communities, including bacteria, archaea, microalgae, and fungi. In some cases, these microbial associates comprise as much as 40% of the sponge volume and can contribute significantly to host metabolism (e.g., via photosynthesis or nitrogen fixation). We review in detail the diversity of microbes associated with sponges, including extensive 16S rRNA-based phylogenetic analyses which support the previously suggested existence of a sponge-specific microbiota. These analyses provide a suitable vantage point from which to consider the potential evolutionary and ecological ramifications of these widespread, sponge-specific microorganisms. Subsequently, we examine the ecology of sponge-microbe associations, including the establishment and maintenance of these sometimes intimate partnerships, the varied nature of the interactions (ranging from mutualism to host-pathogen relationships), and the broad-scale patterns of symbiont distribution. The ecological and evolutionary importance of sponge-microbe associations is mirrored by their enormous biotechnological potential: marine sponges are among the animal kingdom's most prolific producers of bioactive metabolites, and in at least some cases, the compounds are of microbial rather than sponge origin. We review the status of this important field, outlining the various approaches (e.g., cultivation, cell separation, and metagenomics) which have been employed to access the chemical wealth of sponge-microbe associations.
An Acanthamoeba sp. containing two phylogenetically different bacterial endosymbionts
2007 - Environ. Microbiol., 9: 1604-1609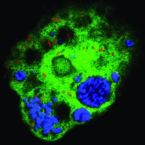
Abstract:
Acanthamoebae are ubiquitous free-living amoebae and important predators of microbial communities. They frequently contain obligate intracellular bacterial symbionts, which show a world-wide distribution. All Acanthamoeba spp. described so far harboured no or only a single specific endosymbiont phylotype, and in some cases evidence for co-evolution between the symbiotic bacteria and the amoeba host has been reported. In this study we have isolated and characterized an Acanthamoeba sp. (strain OEW1) showing a stable symbiotic relationship with two morphologically different endosymbionts. 16S rRNA sequence analysis assigned these symbionts to the candidate genus Procabacter (Betaproteobacteria) and the genus Parachlamydia (Chlamydiae), respectively. Fluorescence in situ hybridization and transmission electron microscopy confirmed the affiliation of the endosymbionts and showed their co-occurrence in the amoeba host cells and their intracellular location within separate compartments enclosed by host-derived membranes. Further analysis of this stable relationship should provide novel insights into the complex interactions of intracellular multiple-partner associations.
Improved 16S rRNA-targeted oligonucleotide probe set for analysis of sulfate-reducing bacteria by fluorescence in situ hybridization
2007 - J. Microbiol. Methods, 69: 523-528
Abstract:
An updated dataset of in silico specificities for 54 previously published 16S rRNA-targeted oligonucleotides was assembled to provide guidance for reliable fluorescence in situ hybridization (FISH) analysis of sulfate-reducing bacteria. Additionally, six new FISH probes were developed for major deltaproteobacterial taxa, including a probe trio targeting most Deltaproteobacteria and Gemmatimonadetes.
Diversity of sulfate-reducing bacteria from an extreme hypersaline sediment in Great Salt Lake (Utah, USA)
2007 - FEMS Microbiol. Ecol., 60: 287-298
Abstract:
The diversity of sulfate-reducing bacteria (SRB) inhabiting the extreme hypersaline sediment (270 g L(-1) NaCl) of the northern arm of Great Salt Lake was studied by integrating cultivation and genotypic identification approaches involving PCR-based retrieval of 16S rRNA and dsrAB genes, the latter encoding major subunits of dissimilatory (bi) sulfite reductase. The majority (85%) of dsrAB sequences retrieved directly from the sediment formed a lineage of high (micro) diversity affiliated with the genus Desulfohalobium, while others represented novel lineages within the families Desulfohalobiaceae and Desulfobacteraceae or among Gram-positive SRB. Using the same sediment, SRB enrichment cultures were established in parallel at 100 and at 190 g L(-1) NaCl using different electron donors. After 5-6 transfers, dsrAB and 16S rRNA gene-based profiling of these enrichment cultures recovered a SRB community composition congruent with the cultivation-independent profiling of the sediment. Pure culture representatives of the predominant Desulfohalobium-related lineage and of one of the Desulfobacteraceae-affilated lineages were successfully obtained. The growth performance of these isolates and of the enrichment cultures suggests that the sediment SRB community of the northern arm of Great Salt Lake consists of moderate halophiles, which are salt-stressed at the in situ salinity of 27%.
Unravelling microbial communities with DNA-microarrays: challenges and future directions
2007 - Microb. Ecol., 53: 498-506
Quantification of uncultured microorganisms by fluorescence microscopy and digital image analysis
2007 - Appl. Microbiol. Biotech., 75: 237-248
Abstract:
Traditional cultivation-based methods to quantify microbial abundance are not suitable for analyses of microbial communities in environmental or medical samples, which consist mainly of uncultured microorganisms. Recently, different cultivation-independent quantification approaches have been developed to overcome this problem. Some of these techniques use specific fluorescence markers, for example ribosomal ribonucleic acid targeted oligonucleotide probes, to label the respective target organisms. Subsequently, the detected cells are visualized by fluorescence microscopy and are quantified by direct visual cell counting or by digital image analysis. This article provides an overview of these methods and some of their applications with emphasis on (semi-)automated image analysis solutions.
Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea
2007 - Environ. Microbiol., 9: 131-142
Abstract:
The Black Sea, with its highly sulfidic water column, is the largest anoxic basin in the world. Within its sediments, the mineralization of organic matter occurs essentially through sulfate reduction and methanogenesis. In this study, the sulfate-reducing community was investigated in order to understand how these microorganisms are distributed relative to the chemical zonation: in the upper sulfate zone, at the sulfate-methane transition zone, and deeply within the methane zone. Total bacteria were quantified by real-time PCR of 16S rRNA genes whereas sulfate-reducing microorganisms (SRM) were quantified by targeting their metabolic key gene, the dissimilatory (bi)sulfite reductase (dsrA). Sulfate-reducing microorganisms were predominant in the sulfate zone but occurred also in the methane zone, relative proportion was maximal around the sulfate-methane transition, c. 30%, and equally high in the sulfate and methane zones, 5-10%. The dsrAB clone library from the sulfate-methane transition zone, showed mostly sequences affiliated with the Desulfobacteraceae. While, the dsrAB clone libraries from the upper, sulfate-rich zone and the deep, sulfate-poor zone were dominated by similar, novel deeply branching sequences which might represent Gram-positive spore-forming sulfate- and/or sulfite-reducing microorganisms. We thus hypothesize that terminal carbon mineralization in surface sediments of the Black Sea is largely due to the sulfate reduction activity of previously hidden SRM. Although these novel SRM were also abundant in sulfate-poor, methanogenic areas of the Black Sea sediment, their activities and possibly very versatile metabolic capabilities remain subject of further study.
probeBase - an online resource for rRNA-targeted oligonucleotide probes: new features 2007
2007 - Nucleic Acids Res., 35: D800-D804
Abstract:
probeBase is a curated database of annotated rRNA-targeted oligonucleotide probes and supporting information. Rapid access to probe, microarray and reference data is achieved by powerful search tools and via different lists that are based on selected categories such as functional or taxonomic properties of the target organism(s), or the hybridization format (fluorescence in situ hybridization or microarray) in which the probes were applied. Additional information on probe coverage and specificity is available through direct submissions of probe sequences from probeBase to RDP-II and Greengenes, two major rRNA sequence databases. A freely-editable user comments field for each probe entry allows any user to add, modify, or remove information, or to report errors in real-time. probeBase entries increased from 700 to more than 1,200 during the past three years. Several options for submission of single probes or entire probe sets, even prior to publication of newly developed probes, should further contribute to keeping probeBase an up-to-date and useful resource. probeBase is freely accessible at http://www.microbial-ecology.net/probebase. Email correspondence can be addressed to probebase.dome@univie.ac.at.
A vista for microbial ecology and environmental biotechnology
2006 - Environ. Sci. Technol., 40: 1096-103
Grazing of a common species of soil protozoa (Acanthamoeba castellanii) affects rhizosphere bacterial community composition and root architecture of rice (Oryza sativa L.)
2006 - Soil Biol. Biochem., 38: 1665-1672Abstract:
We performed a controlled experiment with rice seedlings (Oryza sativa L.) growing in Petri dishes on homogeneous nutrient agar containing a simple rhizosphere food web consisting of a diverse bacterial community and a common soil protozoa, Acanthamoeba castellanii, as bacterial grazer. Presence of amoebae increased bacterial activity and significantly changed the community composition and spatial distribution of bacteria in the rhizosphere. In particular, Betaproteobacteria did benefit from protozoan grazing. We hypothesize that the changes in bacterial community composition affected the root architecture of rice plants. These effects on root architecture affect a fundamental aspect of plant productivity. Root systems in presence of protozoa were characterized by high numbers of elongated (L-type) laterals, those laterals that are a prerequisite for the construction of branched root systems. This was in sharp contrast to root system development in absence of protozoa, where high numbers of lateral root primordia and short (S-type) laterals occurred which did not grow out of the rhizosphere region of the axile root. As a consequence of nutrient release from grazed bacteria and changes in root architecture, the nitrogen content of rice shoots increased by 45% in presence of protozoa. Our study illustrates that interactions over three trophic levels, i.e. between plants, bacteria and protozoa significantly modify root architecture and nutrient uptake by plants.
Wastewater treatment: a model system for microbial ecology
2006 - Trends Biotechnol., 24: 483-489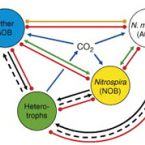
Abstract:
Biological wastewater treatment is among the most important biotechnological applications and, as drivers of the key processes, microorganisms are central to its success. Therefore, the study of wastewater microorganisms has obvious applied significance; however, the importance of wastewater treatment reactors as model systems for microbial ecology is often overlooked. Modern molecular techniques, including environmental genomics, have identified unexpected microbial key players for nutrient removal and sludge bulking and/or foaming, and provided many exciting insights into the diversity, functions and niche differentiations of these predominantly uncultivated microorganisms. It is now time for wastewater microbiology to be recognized as a mature and dynamic discipline in its own right, offering much toward a deeper understanding of life in complex microbial communities. Here, we consider selected key findings to illustrate the past and future roles of molecular ecophysiology and genomics in the development of wastewater microbiology as an important subdiscipline of microbial ecology.
Ecophysiology and niche differentiation of Nitrospira-like bacteria, the key nitrite oxidizers in wastewater treatment plants
2006 - Water Sci. Tech., 54: 21-27
Abstract:
Nitrite-oxidizing bacteria of the genus Nitrospira are key nitrifiers in wastewater treatment plants. Pure cultures of these organisms are unavailable, but cultivation-independent molecular methods make it possible to detect Nitrospira-like bacteria in environmental samples and to investigate their ecophysiology. Comprehensive screening of natural and engineered habitats and of public databases for 16S rRNA sequences of Nitrospira-like bacteria revealed a surprisingly high biodiversity in the genus Nitrospira, which comprises at least four phylogenetic sublineages. All Nitrospira-like bacteria detected in wastewater treatment plants belonged to the sublineages I and II. Subsequently, the population dynamics of different Nitrospira-like bacteria were monitored, by quantitative fluorescence in situ hybridization with rRNA-targeted probes, confocal laser scanning microscopy and digital image analysis, during incubation of nitrifying activated sludge in media containing different nitrite concentrations. These experiments showed that Nitrospira-like bacteria, which were affiliated with the phylogenetic sublineages I or II of the genus Nitrospira, responded differently to nitrite concentration shifts. Previously unknown properties of Nitrospira-like bacteria were discovered in the course of an environmental genomics project. Implications of the obtained results for fundamental understanding of the microbial ecology of nitrite oxidizers as well as for future improvement of nutrient removal in wastewater treatment plants are discussed.
Nitrite concentration influences the population structure of Nitrospira-like bacteria
2006 - Environ. Microbiol., 8: 1487-1495
Abstract:
Chemolithoautotrophic nitrite oxidizers of the genus Nitrospira are a monophyletic but diverse group of organisms, are widely distributed in many natural habitats, and play a key role in nitrogen elimination during biological wastewater treatment. Phylogenetic analyses of cloned 16S rRNA genes and fluorescence in situ hybridization with newly developed rRNA-targeted oligonucleotide probes revealed coexistence of uncultured members of sublineages I and II of the genus Nitrospira in biofilm and activated sludge samples taken from nitrifying wastewater treatment plants. Quantitative microscopic analyses of their spatial arrangement relative to ammonia oxidizers in the biofilm and activated sludge flocs showed that members of the Nitrospira sublineage I occurred significantly more often in immediate vicinity to ammonia oxidizers than would be expected from random community assembly while such a relationship was not observed for Nitrospira sublineage II. This spatial distribution suggested a niche differentiation of these coexisting Nitrospira populations with respect to their preferred concentrations of nitrite. This hypothesis was tested by mathematical modelling of nitrite consumption and resulting nitrite gradients in nitrifying biofilms and by quantifying the abundance of sublineage I and II Nitrospira in activated sludge during incubations with nitrite in different concentrations. Consistent with the observed localization patterns, a higher nitrite concentration selected for sublineage I but suppressed sublineage II Nitrospira.
Tapping the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia amoebophila
2006 - Mol. Microbiol., 60: 1534-1545
Abstract:
Protochlamydia amoebophila UWE25 is related to the Chlamydiaceae comprising major pathogens of humans, but thrives as obligate intracellular symbiont in the protozoan host Acanthamoeba sp. The genome of P. amoebophila encodes five paralogous carrier proteins belonging to the nucleotide transporter (NTT) family. Here we report on three P. amoebophila NTT isoforms, PamNTT2, PamNTT3, and PamNTT5, which possess several conserved amino acid residues known to be critical for nucleotide transport. We demonstrated that these carrier proteins are able to transport nucleotides, although substrate specificities and mode of transport differ in an unexpected manner and are unique among known NTTs. PamNTT2 is a counter exchange transporter exhibiting sub-millimolar apparent affinities for all four RNA nucleotides, PamNTT3 catalyses an unidirectional proton-coupled transport confined to UTP, whereas PamNTT5 mediates a proton-energized GTP and ATP import. All NTT genes of P. amoebophila are transcribed during intracellular multiplication in acanthamoebae. The biochemical characterization of all five NTT proteins from P. amoebophila in this and previous studies uncovered that these metabolically impaired bacteria are intimately connected with their host cell’s metabolism in a surprisingly complex manner.
The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance.
2006 - Curr. Opin. Biotechnol., 3: 241-9Abstract:
In the rRNA-based tree of life four bacterial phyla, comprising the Planctomycetes, Verrucomicrobia, Chlamydiae and Lentisphaerae, form together with the candidate phyla Poribacteria and OP3 a monophyletic group referred to as the PVC superphylum. This assemblage contains organisms that possess dramatically different lifestyles and which colonize sharply contrasting habitats. Some members of this group are among the most successful human pathogens, others are abundant soil microbes, and others still are of major importance for the marine nitrogen cycle and hold much promise for sustainable wastewater treatment. Recent comparative genomic and metagenomic analyses of a few representatives of this group revealed many unusual features and generated unexpected hypotheses regarding their physiology, some of which have already been confirmed experimentally. Furthermore, the availability of these genome sequences offered new insights into the evolutionary history of this peculiar group of microbes with major medical, ecological and biotechnological relevance.
Deciphering the evolution and metabolism of an anammox bacterium from a community genome
2006 - Nature, 440: 790-794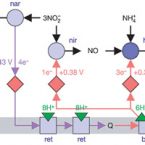
Abstract:
Anaerobic ammonium oxidation (anammox) has become a main focus in oceanography and wastewater treatment. It is also the nitrogen cycle's major remaining biochemical enigma. Among its features, the occurrence of hydrazine as a free intermediate of catabolism, the biosynthesis of ladderane lipids and the role of cytoplasm differentiation are unique in biology. Here we use environmental genomics the reconstruction of genomic data directly from the environment to assemble the genome of the uncultured anammox bacterium Kuenenia stuttgartiensis from a complex bioreactor community. The genome data illuminate the evolutionary history of the Planctomycetes and allow us to expose the genetic blueprint of the organism's special properties. Most significantly, we identified candidate genes responsible for ladderane biosynthesis and biological hydrazine metabolism, and discovered unexpected metabolic versatility.
Non-sulfate-reducing, syntrophic bacteria affiliated with the Desulfotomaculum cluster I are widely distributed in methanogenic environments
2006 - Appl. Environ. Microbiol., 72: 2080-2091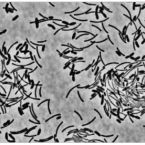
Abstract:
The classical perception of members of the gram-positive Desulfotomaculum cluster I as sulfate-reducing bacteria was recently challenged by the isolation of new representatives lacking the ability for anaerobic sulfate respiration. For example, the two described syntrophic propionate-oxidizing species of the genus Pelotomaculum form the novel Desulfotomaculum subcluster Ih. In the present study, we applied a polyphasic approach by using cultivation-independent and culturing techniques in order to further characterize the occurrence, abundance, and physiological properties of subcluster Ih bacteria in low-sulfate, methanogenic environments. 16S rRNA (gene)-based cloning, quantitative fluorescence in situ hybridization, and real-time PCR analyses showed that the subcluster Ih population composed a considerable part of the Desulfotomaculum cluster I community in almost all samples examined. Additionally, five propionate-degrading syntrophic enrichments of subcluster Ih bacteria were successfully established, from one of which the new strain MGP was isolated in coculture with a hydrogenotrophic methanogen. None of the cultures analyzed, including previously described Pelotomaculum species and strain MGP, consumed sulfite, sulfate, or organosulfonates. In accordance with these phenotypic observations, a PCR-based screening for dsrAB (key genes of the sulfate respiration pathway encoding the alpha and beta subunits of the dissimilatory sulfite reductase) of all enrichments/(co)cultures was negative with one exception. Surprisingly, strain MGP contained dsrAB, which were transcribed in the presence and absence of sulfate. Based on these and previous findings, we hypothesize that members of Desulfotomaculum subcluster Ih have recently adopted a syntrophic lifestyle to thrive in low-sulfate, methanogenic environments and thus have lost their ancestral ability for dissimilatory sulfate/sulfite reduction.
Candidatus Thiobios zoothamnicoli, an ectosymbiotic bacterium covering the giant marine ciliate Zoothamnium niveum
2006 - Appl. Environ. Microbiol., 72: 2014-2021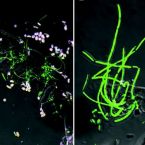
Abstract:
Zoothamnium niveum is a giant, colonial marine ciliate from sulfide-rich habitats obligatorily covered with chemoautotrophic, sulfide-oxidizing bacteria which appear as coccoid rods and rods with a series of intermediate shapes. Comparative 16S rRNA gene sequence analysis and fluorescence in situ hybridization showed that the ectosymbiont of Z. niveum belongs to only one pleomorphic phylotype. The Z. niveum ectosymbiont is only moderately related to previously identified groups of thiotrophic symbionts within the Gammaproteobacteria, and shows highest 16S rRNA sequence similarity with the free-living sulfur-oxidizing bacterial strain ODIII6 from shallow-water hydrothermal vents of the Mediterranean Sea (94.5%) and an endosymbiont from a deep-sea hydrothermal vent gastropod of the Indian Ocean Ridge (93.1%). A replacement of this specific ectosymbiont by a variety of other bacteria was observed only for senescent basal parts of the host colonies. The taxonomic status "Candidatus Thiobios zoothamnicoli" is proposed for the ectosymbiont of Z. niveum based on its ultrastructure, its 16S rRNA gene, the intergenic spacer region, and its partial 23S rRNA gene sequence.
Selective enrichment and molecular characterization of a previously uncultured Nitrospira-like bacterium from activated sludge
2006 - Environ. Microbiol., 8: 405-415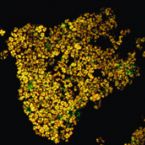
Abstract:
Previously uncultured nitrite-oxidizing bacteria affiliated to the genus Nitrospira have for the first time been successfully enriched from activated sludge from a municipal wastewater treatment plant. During the enrichment procedure, the abundance of the Nitrospira-like bacteria increased to approximately 86% of the total bacterial population. This high degree of purification was achieved by a novel enrichment protocol, which exploits physiological features of Nitrospira-like bacteria and includes the selective repression of coexisting Nitrobacter cells and heterotrophic contaminants by application of ampicillin in a final concentration of 50 microg ml(-1). The enrichment process was monitored by electron microscopy, fluorescence in situ hybridization (FISH) with rRNA-targeted probes and fatty acid profiling. Phylogenetic analysis of 16S rRNA gene sequences revealed that the enriched bacteria represent a novel Nitrospira species closely related to uncultured Nitrospira-like bacteria previously found in wastewater treatment plants and nitrifying bioreactors. The enriched strain is provisionally classified as 'Candidatus Nitrospira defluvii'.
Cohn's Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase
2006 - Proc. Natl. Acad. Sci. USA, 7: 2363-2367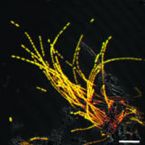
Abstract:
135 years ago Ferdinand Cohn, the founder of bacteriology, microscopically observed a conspicuous filamentous bacterium with a complex life cycle and described it as Crenothrix polyspora. This uncultured bacterium is infamous for mass developments in drinking water systems, but its phylogeny and physiology remained unknown. We show that C. polyspora is a gammaproteobacterium closely related to methanotrophs and capable of oxidizing methane. We discovered that C. polyspora encodes a phylogenetically very unusual particulate methane monooxygenase whose expression is strongly increased in the presence of methane. Our findings demonstrate a previously unrecognized complexity of the evolutionary history and cell biology of methane-oxidizing bacteria.
daime, a novel image analysis program for microbial ecology and biofilm research
2006 - Environ. Microbiol., 8: 200-213
Abstract:
Combinations of microscopy and molecular techniques to detect, identify and characterize microorganisms in environmental and medical samples are widely used in microbial ecology and biofilm research. The scope of these methods, which include fluorescence in situ hybridization (FISH) with rRNA-targeted probes, is extended by digital image analysis routines that extract from micrographs important quantitative data. Here we introduce daime (digital image analysis in microbial ecology), a new computer program integrating 2-D and 3-D image analysis and visualization functionality, which has previously not been available in a single open-source software package. For example, daime automatically finds 2-D and 3-D objects in images and confocal image stacks, and offers special functions for quantifying microbial populations and evaluating new FISH probes. A novel feature is the quantification of spatial localization patterns of microorganisms in complex samples like biofilms. In combination with '3D-FISH', which preserves the 3-D structure of samples, this stereological technique was applied in a proof of principle experiment on activated sludge and provided quantitative evidence that functionally linked ammonia and nitrite oxidizers cluster together in their habitat. This image analysis method complements recent molecular techniques for analysing structure-function relationships in microbial communities and will help to characterize symbiotic interactions among microorganisms.
Linking microbial community structure with function: fluorescence in situ hybridization-microautoradiography and isotope arrays
2006 - Curr. Opin. Biotechnol., 17: 1-9
Abstract:
The ecophysiology of microorganisms has been at the heart of microbial ecology since its early days, but only during the past decade have methods become available for cultivation-independent, direct identification of microorganisms in complex communities and for the simultaneous investigation of their activity and substrate uptake patterns. The combination of fluorescence in situ hybridization (FISH) and microautoradiography (MAR) is currently the most widely applied tool for revealing physiological properties of microorganisms in their natural environment with single-cell resolution. For example, this technique has been used in wastewater treatment and marine systems to describe the functional properties of newly discovered species, and to identify microorganisms responsible for key physiological processes. Recently, the scope of FISH-MAR was extended by rendering it quantitative and by combining it with microelectrode measurements or stable isotope probing. Isotope arrays have also been developed that exploit the parallel detection offered by DNA microarrays to measure incorporation of labelled substrate into the rRNA of many community members in a single experiment.
Functional marker genes for identification of sulphate-reducing prokaryotes
2005 - Methods Enzymol., 397: 469-489Abstract:
Sulfate-reducing prokaryotes (SRPs) exploit sulfate as an electron acceptor for anaerobic respiration and exclusively catalyze this essential step of the world's sulfur cycle. Because SRPs are found in many prokaryotic phyla and are often closely related to non-SRPs, 16S rRNA gene-based analyses are inadequate to identify novel lineages of this guild in a cultivation-independent manner. This problem can be solved by comparative sequence analysis of environmentally retrieved gene fragments of the dissimilatory (bi)sulfite (dsrAB) and adenosine-5'-phosphosulfate reductases (apsA), which encode key enzymes of the SRP energy metabolism. This chapter provides detailed protocols for the application of these functional marker molecules for SRP diversity surveys in the environment. Data from the analysis of dsrAB sequence diversity in water samples from the Mariager Fjord in northeast Denmark are presented to illustrate the different steps of the protocols. Furthermore, this chapter describes a novel gel retardation-based technique, suitable for fingerprinting of the approximately 1.9-kb-large dsrAB polymerase chain reaction amplification products, which efficiently increases the chance of retrieving rare and novel dsrAB sequence types from environmental samples.
'Candidatus Protochlamydia amoebophila', an endosymbiont of Acanthamoeba spp
2005 - Int. J. Syst. Evol. Microbiol., 55: 1863-1866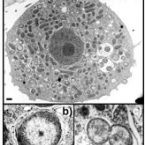
Abstract:
The obligately intracellular coccoid bacterium UWE25, a symbiont of Acanthamoeba spp., was previously identified as being related to chlamydiae based upon the presence of a chlamydia-like developmental cycle and its 16S rRNA gene sequence. Analysis of its complete genome sequence demonstrated that UWE25 shows many characteristic features of chlamydiae, including dependency on host-derived metabolites, composition of the cell envelope and the ability to thrive as an energy parasite within the cells of its eukaryotic host. Phylogenetic analysis of 44 ribosomal proteins further confirmed the affiliation of UWE25 to the 'Chlamydiae'. Within this phylum, UWE25 could be assigned to the family Parachlamydiaceae based on comparative analyses of the 16S rRNA, 23S rRNA and endoribonuclease P RNA genes. The distinct dissimilarities from its closest relative, Parachlamydia acanthamoebae Bn(9)(T) (7.1, 9.7 and 28.8%, respectively), observed in this analysis justify its classification in a new genus. Therefore, the name 'Candidatus Protochlamydia amoebophila' is proposed for the designation of the Acanthamoeba sp. symbiont UWE25 (=ATCC PRA-7).
New insights into metabolic properties of marine bacteria encoding proteorhodopsins
2005 - PLoS Biol., 3: e273
Abstract:
Proteorhodopsin phototrophy was recently discovered in oceanic surface waters. In an effort to characterize uncultured proteorhodopsin-exploiting bacteria, large-insert bacterial artificial chromosome (BAC) libraries from the Mediterranean Sea and Red Sea were analyzed. Fifty-five BACs carried diverse proteorhodopsin genes, and we confirmed the function of five. We calculate that proteorhodopsin-exploiting bacteria account for 13% of microorganisms in the photic zone. We further show that some proteorhodopsin-containing bacteria possess a retinal biosynthetic pathway and a reverse sulfite reductase operon, employed by prokaryotes oxidizing sulfur compounds. Thus, these novel phototrophs are an unexpectedly large and metabolically diverse component of the marine microbial surface water.
Oligonucleotide microarray for identification of Enterococcus species
2005 - FEMS Microbiol. Lett., 246: 133-142
Abstract:
For detection of most members of the Enterococcaceae, the specificity of a novel oligonucleotide microarray (ECC-PhyloChip) consisting of 41 hierarchically nested 16S or 23S rRNA gene-targeted probes was evaluated with 23 pure cultures (including 19 Enterococcus species). Target nucleic acids were prepared by PCR amplification of a 4.5-kb DNA fragment containing large parts of the 16S and 23S rRNA genes and were subsequently labeled fluorescently by random priming. Each tested member of the Enterococcaceae was correctly identified on the basis of its unique microarray hybridization pattern. The evaluated ECC-PhyloChip was successfully applied for identification of Enterococcus faecium and Enterococcus faecalis in artificially contaminated milk samples demonstrating the utility of the ECC-PhyloChip for parallel identification and differentiation of Enterococcus species in food samples.
Biomarkers for in situ detection of anaerobic ammonium-oxidizing (ANAMMOX) bacteria
2005 - Appl. Environ. Microbiol., 71: 1677-1684
Malikia granosa gen. nov., sp. nov., a novel polyhydroxyalkanoate- and polyphosphate-accumulating bacterium isolated from activated sludge and reclassification of Pseudomonas spinosa as Malikia spinosa comb. nov
2005 - Int. J. Syst. Evol. Microbiol., 55: 621-629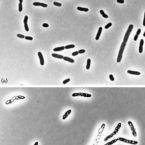
Abstract:
A Gram-negative, motile, rod-shaped bacterium, designated strain P1(T), was isolated from activated sludge of a municipal wastewater treatment plant. Phylogenetic analysis of its 16S rRNA gene sequence placed the novel isolate among representatives of the family Comamonadaceae. The closest relatives in reconstructed phylogenetic trees were Pseudomonas spinosa, Macromonas bipunctata and Hydrogenophaga species. Strain P1(T) was not able to grow anaerobically or autotrophically, reduced nitrate to nitrite and required vitamins for growth. Ubiquinone 8 (Q8) and 3-hydroxy-substituted fatty acids were present, but 2-hydroxy fatty acids were absent. The G+C content of the DNA was 67 mol%. Phenotypic characteristics allowed a clear differentiation of strain P1(T) from representatives of the genera Hydrogenophaga and Macromonas, whereas DNA-DNA hybridization experiments revealed that strain P1(T) did not belong to the species P. spinosa. As a peculiarity, cells of strain P1(T) and P. spinosa ATCC 14606(T) were able to accumulate large amounts of polyhydroxyalkanoates and polyphosphate in the form of large intracellular granules. Apparently in both strains nitrogen limitation stimulates the production of polyhydroxyalkanoates, whereas carbon starvation induces the formation of polyphosphates. Based upon phylogenetic and phenotypic evidence, it is proposed to establish the novel taxon Malikia granosa gen. nov., sp. nov., represented by the type strain P1(T) (=DSM 15619(T)=JCM 12706(T)=CIP 108194(T)). The most closely related species of strain P1(T) was P. spinosa. This species has been misclassified, and it is proposed to transfer it to the new genus Malikia as Malikia spinosa gen. nov., comb. nov. The type strain is ATCC 14606(T) (=DSM 15801(T)).
16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order Rhodocyclales
2005 - Appl. Environ. Microbiol., 71: 1373-1386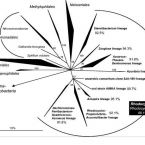
Abstract:
For simultaneous identification of members of the betaproteobacterial order Rhodocyclales in environmental samples, a 16S rRNA gene-targeted oligonucleotide microarray (RHC-PhyloChip) consisting of 79 probes was developed. Probe design was based on phylogenetic analysis of available 16S rRNA sequences from all cultured and as yet uncultured members of the Rhodocyclales. The multiple nested probe set was evaluated for microarray hybridization with 16S rRNA gene PCR amplicons from 29 reference organisms. Subsequently, the RHC-PhyloChip was successfully used for cultivation-independent Rhodocyclales diversity analysis in activated sludge from an industrial wastewater treatment plant. The implementation of a newly designed Rhodocyclales-selective PCR amplification system prior to microarray hybridization greatly enhanced the sensitivity of the RHC-PhyloChip and thus enabled the detection of Rhodocyclales populations with relative abundances of less than 1% of all bacteria (as determined by fluorescence in situ hybridization) in the activated sludge. The presence of as yet uncultured Zoogloea-, Ferribacterium/Dechloromonas-, and Sterolibacterium-related bacteria in the industrial activated sludge, as indicated by the RHC-PhyloChip analysis, was confirmed by retrieval of their 16S rRNA gene sequences and subsequent phylogenetic analysis, demonstrating the suitability of the RHC-PhyloChip as a novel monitoring tool for environmental microbiology.
Lateral gene transfer of dissimilatory (bi)sulfite reductase revisited
2005 - J. Bacteriol., 187: 2203-2208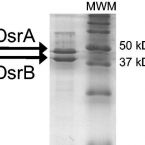
Evolutionary history of the genus Listeria and its virulence genes
2005 - Syst. Appl. Microbiol., 28: 1-18Abstract:
The genus Listeria contains the two pathogenic species Listeria monocytogenes and Listeria ivanovii and the four apparently apathogenic species Listeria innocua, Listeria seeligeri, Listeria welshimeri, and Listeria grayi. Pathogenicity of the former two species is enabled by an approximately 9 kb virulence gene cluster which is also present in a modified form in L. seeligeri. For all Listeria species, the sequence of the virulence gene cluster locus and its flanking regions was either determined in this study or assembled from public databases. Furthermore, some virulence-associated internalin loci were compared among the six species. Phylogenetic analyses were performed on a data set containing the sequences of prs, ldh, vclA, and vclB (all directly flanking the virulence gene cluster), as well as the iap gene and the 16S and 23S-rRNA coding genes which are located at different sites in the listerial chromosomes. L. grayi represents the deepest branch within the genus. The remaining five species form two groupings which have a high bootstrap support and which are consistently found by using different treeing methods. One lineage represents L. monocytogenes and L. innocua, while the other contains L. welshimeri, L. ivanovii and L. seeligeri, with L. welshimeri forming the deepest branch. Based on this perception, we tried to reconstruct the evolution of the virulence gene cluster. Since no traces of lateral gene transfer events could be detected the most parsimonious scenario is that the virulence gene cluster was present in the common ancestor of L. monocytogenes, L. innocua, L. ivanovii, L. seeligeri and L. welshimeri and that the pathogenic capability has been lost in two separate events represented by L. innocua and L. welshimeri. This hypothesis is also supported by the location of the putative deletion breakpoints of the virulence gene cluster within L. innocua and L. welshimeri.
Amoeba as training grounds intracellular bacterial pathogens
2005 - Appl. Environ. Microbiol., 71: 20-28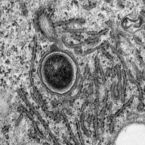
Recovery of an environmental chlamydia strain from activated sludge by co-cultivation with Acanthamoeba sp
2005 - Microbiology, 151: 301-309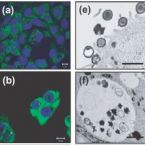
Abstract:
Chlamydiae are a unique group of obligate intracellular bacteria comprising important pathogens of vertebrates as well as symbionts of free-living amoebae. Although there is ample molecular evidence for a huge diversity and wide distribution of chlamydiae in nature, environmental chlamydiae are currently represented by only few isolates. This paper reports the recovery of a novel environmental chlamydia strain from activated sludge by co-cultivation with Acanthamoeba sp. The recovered environmental chlamydia strain UV-7 showed the characteristic morphology of chlamydial developmental stages as revealed by electron microscopy and was identified as a new member of the family Parachlamydiaceae (98.7 % 16S rRNA sequence similarity to Parachlamydia acanthamoebae). Infection studies suggested that Parachlamydia sp. UV-7 is not confined to amoeba hosts but is also able to invade mammalian cells. These findings outline a new straightforward approach to retrieving environmental chlamydiae from nature without prior, tedious isolation and cultivation of their natural host cells, and lend further support to suggested implications of environmental chlamydiae for public health.
Microarray and functional gene analyses of sulfate-reducing prokaryotes in low-sulfate, acidic fens reveal cooccurrence of recognized genera and novel lineages.
2004 - Appl. Environ. Microbiol., 70: 6998-7009
Abstract:
Low-sulfate, acidic (approximately pH 4) fens in the Lehstenbach catchment in the Fichtelgebirge mountains in Germany are unusual habitats for sulfate-reducing prokaryotes (SRPs) that have been postulated to facilitate the retention of sulfur and protons in these ecosystems. Despite the low in situ availability of sulfate (concentration in the soil solution, 20 to 200 microM) and the acidic conditions (soil and soil solution pHs, approximately 4 and 5, respectively), the upper peat layers of the soils from two fens (Schlöppnerbrunnen I and II) of this catchment displayed significant sulfate-reducing capacities. 16S rRNA gene-based oligonucleotide microarray analyses revealed stable diversity patterns for recognized SRPs in the upper 30 cm of both fens. Members of the family "Syntrophobacteraceae" were detected in both fens, while signals specific for the genus Desulfomonile were observed only in soils from Schlöppnerbrunnen I. These results were confirmed and extended by comparative analyses of environmentally retrieved 16S rRNA and dissimilatory (bi)sulfite reductase (dsrAB) gene sequences; dsrAB sequences from Desulfobacca-like SRPs, which were not identified by microarray analysis, were obtained from both fens. Hypotheses concerning the ecophysiological role of these three SRP groups in the fens were formulated based on the known physiological properties of their cultured relatives. In addition to these recognized SRP lineages, six novel dsrAB types that were phylogenetically unrelated to all known SRPs were detected in the fens. These dsrAB sequences had no features indicative of pseudogenes and likely represent novel, deeply branching, sulfate- or sulfite-reducing prokaryotes that are specialized colonists of low-sulfate habitats.
Deciphering the function of uncultured microorganisms
2004 - ASM News, 70: 63-70
Chlamydial endocytobionts of free-living amoebae differentially affect the growth rate of their hosts
2004 - Europ. J. Protist., 40: 57-60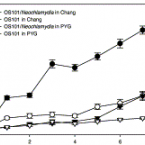
A candidate NAD+ transporter in an intracellular bacterial symbiont related to chlamydiae
2004 - Nature, 432: 622-625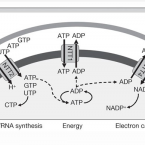
Abstract:
Bacteria living within eukaryotic cells can be essential for the survival or reproduction of the host but in other cases are among the most successful pathogens. Environmental Chlamydiae, including strain UWE25, thrive as obligate intracellular symbionts within protozoa; are recently discovered relatives of major bacterial pathogens of humans; and also infect human cells. Genome analysis of UWE25 predicted that this symbiont is unable to synthesize the universal electron carrier nicotinamide adenine dinucleotide (NAD+). Compensation of limited biosynthetic capacity in intracellular bacteria is usually achieved by import of primary metabolites. Here, we report the identification of a candidate transporter protein from UWE25 that is highly specific for import of NAD+ when synthesized heterologously in Escherichia coli. The discovery of this candidate NAD+/ADP exchanger demonstrates that intact NAD+ molecules can be transported through cytoplasmic membranes. This protein acts together with a newly discovered nucleotide transporter and an ATP/ADP translocase, and allows UWE25 to exploit its host cell by means of a sophisticated metabolic parasitism.
Bacterial endosymbionts of free-living amoebae
2004 - J. Euk. Microbiol., 51: 509-514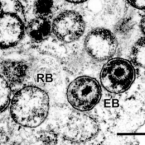
Abstract:
The occurrence of bacterial endosymbionts in free-living amoebae has been known for decades, but their obligate intracellular lifestyle hampered their identification. Only recently, application of the full cycle rRNA approach including 16S rRNA gene sequencing and fluorescence in situ hybridization with 16S rRNA-targeted oligonucleotide probes assigned the symbionts of Acanthamoeba spp. and Hartmannella sp. to five different evolutionary lineages within the Proteobacteria, the Bacteroidetes, and the Chlamydiae, respectively. Some of these bacterial symbionts are most closely related to bacterial pathogens of humans, and it has been suggested that they should be considered potential emerging pathogens. Complete genome sequence analysis of a chlamydia-related symbiont of Acanthamoeba sp. showed that this endosymbiont uses similar mechanisms for interaction with its eukaryotic host cell as well known bacterial pathogens of humans. Furthermore, phylogenetic analysis suggested that these mechanisms have been invented by the ancestor of the amoeba symbionts in interplay with ancient unicellular eukaryotes.
Discovery of the novel candidate phylum "Poribacteria" in marine sponges
2004 - Appl. Environ. Microbiol., 70: 3724-3732
Abstract:
Marine sponges (Porifera) harbor large amounts of commensal microbial communities within the sponge mesohyl. We employed 16S rRNA gene library construction using specific PCR primers to provide insights into the phylogenetic identity of an abundant sponge-associated bacterium that is morphologically characterized by the presence of a membrane-bound nucleoid. In this study, we report the presence of a previously unrecognized evolutionary lineage branching deeply in the domain Bacteria that is moderately related to the Planctomycetes, Verrucomicrobia, and Chlamydia lines of decent. Because members of this lineage showed <75% 16S rRNA gene sequence similarity to known bacterial phyla, we suggest the status of a new candidate phylum, named "Poribacteria", to acknowledge the affiliation of the new bacterium with sponges. The affiliation of the morphologically conspicuous sponge bacterium with the novel phylogenetic lineage was confirmed by fluorescence in situ hybridization with newly designed probes targeting different sites of the poribacterial 16S rRNA. Consistent with electron microscopic observations of cell compartmentalization, the fluorescence signals appeared in a ring-shaped manner. PCR screening with "Poribacteria"-specific primers gave positive results for several other sponge species, while samples taken from the environment (seawater, sediments, and a filter-feeding tunicate) were PCR negative. In addition to a report for Planctomycetes, this is the second report of cell compartmentalization, a feature that was considered exclusive to the eukaryotic domain, in prokaryotes.
Illuminating the evolutionary history of chlamydiae
2004 - Science, 304: 728-730- Supplementary material [PDF]
- Press release [PDF]
- Press release (German) [PDF]
- Movie showing environmental chlamydia UWE25 within its amoeba host cells. AAAS/Science. [Windows Media Player]
- Movie showing environmental chlamydia UWE25 within its amoeba host cells. AAAS/Science.[Quicktime]
- Environmental chlamydia genome database
Abstract:
Chlamydiae are the major cause of preventable blindness and sexually transmitted disease. Genome analysis of a chlamydia-related symbiont of free-living amoebae revealed that it is twice as large as any of the pathogenic chlamydiae and had few signs of recent lateral gene acquisition. We showed that about 700 million years ago the last common ancestor of pathogenic and symbiotic chlamydiae was already adapted to intracellular survival in early eukaryotes and contained many virulence factors found in modern pathogenic chlamydiae, including a type III secretion system. Ancient chlamydiae appear to be the originators of mechanisms for the exploitation of eukaryotic cells.
Ottowia thiooxydans gen. nov., sp. nov., a novel facultatively anaerobic, N2O producing bacterium isolated from activated sludge and transfer of Aquaspirillum gracile to Hylemonella gracilis gen. nov. comb. nov
2004 - Int. J. Syst. Evol. Microbiol., 54: 99-106
ATP/ADP translocases: A common feature of obligate intracellular amoebal symbionts related to chlamydiae and rickettsiae
2004 - J. Bacteriol., 186: 683-691
Abstract:
ATP/ADP translocases catalyze the highly specific transport of ATP across a membrane in an exchange mode with ADP. Such unique transport proteins are employed by plant plastids and have among the prokaryotes so far only been identified in few obligate intracellular bacteria belonging to the Chlamydiales and the Rickettsiales. In this study, 12 phylogenetically diverse bacterial endosymbionts of free-living amoebae and paramecia were screened for the presence of genes encoding ATP/ADP transport proteins. The occurrence of ATP/ADP translocase genes was found to be restricted to endosymbionts related to rickettsiae and chlamydiae. We showed that the ATP/ADP transport protein of the Parachlamydia-related endosymbiont of Acanthamoeba sp. strain UWE25, a recently identified relative of the important human pathogens Chlamydia trachomatis and Chlamydophila pneumoniae, is functional when expressed in the heterologous host Escherichia coli and demonstrated the presence of transcripts during the chlamydial developmental cycle. These findings indicate that the interaction between Parachlamydia-related endosymbionts and their amoeba hosts concerns energy parasitism similar to the interaction between pathogenic chlamydiae and their human host cells. Phylogenetic analysis of all known ATP/ADP translocases indicated that the genes encoding ATP/ADP translocases originated from a chlamydial ancestor and were, after an ancient gene duplication, transferred horizontally to rickettsiae and plants.
Use of stable-isotope probing, full-cycle rRNA analysis, and fluorescence in situ hybridization-micrautoradiography to study a methanol-fed denitrifying microbial community
2004 - Appl. Environ. Microbiol., 70: 588-596
Abstract:
A denitrifying microbial consortium was enriched in an anoxically operated, methanol-fed sequencing batch reactor (SBR) fed with a mineral salts medium containing methanol as the sole carbon source and nitrate as the electron acceptor. The SBR was inoculated with sludge from a biological nutrient removal activated sludge plant exhibiting good denitrification. The SBR denitrification rate improved from less than 0.02 mg of NO(3)(-)-N mg of mixed-liquor volatile suspended solids (MLVSS)(-1) h(-1) to a steady-state value of 0.06 mg of NO(3)(-)-N mg of MLVSS(-1) h(-1) over a 7-month operational period. At this time, the enriched microbial community was subjected to stable-isotope probing (SIP) with [(13)C]methanol to biomark the DNA of the denitrifiers. The extracted [(13)C]DNA and [(12)C]DNA from the SIP experiment were separately subjected to full-cycle rRNA analysis. The dominant 16S rRNA gene phylotype (group A clones) in the [(13)C]DNA clone library was closely related to those of the obligate methylotrophs Methylobacillus and Methylophilus in the order Methylophilales of the Betaproteobacteria (96 to 97% sequence identities), while the most abundant clone groups in the [(12)C]DNA clone library mostly belonged to the family Saprospiraceae in the Bacteroidetes phylum. Oligonucleotide probes for use in fluorescence in situ hybridization (FISH) were designed to specifically target the group A clones and Methylophilales (probes DEN67 and MET1216, respectively) and the Saprospiraceae clones (probe SAP553). Application of these probes to the SBR biomass over the enrichment period demonstrated a strong correlation between the level of SBR denitrification and relative abundance of DEN67-targeted bacteria in the SBR community. By contrast, there was no correlation between the denitrification rate and the relative abundances of the well-known denitrifying genera Hyphomicrobium and Paracoccus or the Saprospiraceae clones visualized by FISH in the SBR biomass. FISH combined with microautoradiography independently confirmed that the DEN67-targeted cells were the dominant bacterial group capable of anoxic [(14)C]methanol uptake in the enriched biomass. The well-known denitrification lag period in the methanol-fed SBR was shown to coincide with a lag phase in growth of the DEN67-targeted denitrifying population. We conclude that Methylophilales bacteria are the dominant denitrifiers in our SBR system and likely are important denitrifiers in full-scale methanol-fed denitrifying sludges.
Use of microautoradiography to study in situ physiology of bacteria in biofilms
2003 - Rev. Environ. Sci. Biotechnol., 2: 261-268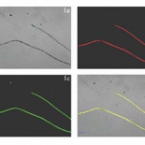
Structure and activity of multiple nitrifying bacterial populations coexisting in a biofilm
2003 - Environ. Microbiol., 5: 355-369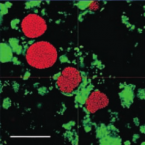
Monitoring microbial diversity and natural products profiles of the sponge Aplysina cavernicola following transplantation
2003 - Marine Biology, 142: 685-692The isotope array, a new tool that employs substrate-mediated labeling of rRNA for determination of microbial community structure and function
2003 - Appl. Environ. Microbiol., 69: 6875-6887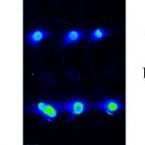
Abstract:
A new microarray method, the isotope array approach, for identifying microorganisms which consume a (14)C-labeled substrate within complex microbial communities was developed. Experiments were performed with a small microarray consisting of oligonucleotide probes targeting the 16S rRNA of ammonia-oxidizing bacteria (AOB). Total RNA was extracted from a pure culture of Nitrosomonas eutropha grown in the presence of [(14)C]bicarbonate. After fluorescence labeling of the RNA and microarray hybridization, scanning of all probe spots for fluorescence and radioactivity revealed that specific signals were obtained and that the incorporation of (14)C into rRNA could be detected unambiguously. Subsequently, we were able to demonstrate the suitability of the isotope array approach for monitoring community composition and CO(2) fixation activity of AOB in two nitrifying activated-sludge samples which were incubated with [(14)C]bicarbonate for up to 26 h. AOB community structure in the activated-sludge samples, as predicted by the microarray hybridization pattern, was confirmed by quantitative fluorescence in situ hybridization (FISH) and comparative amoA sequence analyses. CO(2) fixation activities of the AOB populations within the complex activated-sludge communities were detectable on the microarray by (14)C incorporation and were confirmed independently by combining FISH and microautoradiography. AOB rRNA from activated sludge incubated with radioactive bicarbonate in the presence of allylthiourea as an inhibitor of AOB activity showed no incorporation of (14)C and thus was not detectable on the radioactivity scans of the microarray. These results suggest that the isotope array can be used in a PCR-independent manner to exploit the high parallelism and discriminatory power of microarrays for the direct identification of microorganisms which consume a specific substrate in the environment.
16S rRNA and amoA-based Phylogeny of 12 Novel Betaproteobacterial Ammonia Oxidizing Isolates: Extension of the Data Set and Proposal of a New Lineage within the Nitrosomonads
2003 - Int. J. Syst. Evol. Microbiol., 53: 1485-1494Abstract:
The phylogenetic relationship of 12 ammonia-oxidizing isolates (eight nitrosospiras and four nitrosomonads), for which no gene sequence information was available previously, was investigated based on their genes encoding 16S rRNA and the active site subunit of ammonia monooxygenase (AmoA). Almost full-length 16S rRNA gene sequences were determined for the 12 isolates. In addition, 16S rRNA gene sequences of 15 ammonia-oxidizing bacteria (AOB) published previously were completed to allow for a more reliable phylogeny inference of members of this guild. Moreover, sequences of 453 bp fragments of the amoA gene were determined from 15 AOB, including the 12 isolates, and completed for 10 additional AOB. 16S rRNA gene and amoA-based analyses, including all available sequences of AOB pure cultures, were performed to determine the position of the newly retrieved sequences within the established phylogenetic framework. The resulting 16S rRNA gene and amoA tree topologies were similar but not identical and demonstrated a superior resolution of 16S rRNA versus amoA analysis. While 11 of the 12 isolates could be assigned to different phylogenetic groups recognized within the betaproteobacterial AOB, the estuarine isolate Nitrosomonas sp. Nm143 formed a separate lineage together with three other marine isolates whose 16S rRNA sequences have not been published but have been deposited in public databases. In addition, 17 environmentally retrieved 16S rRNA gene sequences not assigned previously and all originating exclusively from marine or estuarine sites clearly belong to this lineage.
Filamentous Epsilonproteobacteria dominate microbial mats from sulfidic cave springs
2003 - Appl. Environ. Microbiol., 69: 5503-5511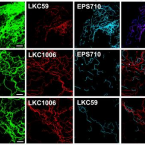
Abstract:
Hydrogen sulfide-rich groundwater discharges from springs into Lower Kane Cave, Wyoming, where microbial mats dominated by filamentous morphotypes are found. The full-cycle rRNA approach, including 16S rRNA gene retrieval and fluorescence in situ hybridization (FISH), was used to identify these filaments. The majority of the obtained 16S rRNA gene clones from the mats were affiliated with the "Epsilonproteobacteria" and formed two distinct clusters, designated LKC group I and LKC group II, within this class. Group I was closely related to uncultured environmental clones from petroleum-contaminated groundwater, sulfidic springs, and sulfidic caves (97 to 99% sequence similarity), while group II formed a novel clade moderately related to deep-sea hydrothermal vent symbionts (90 to 94% sequence similarity). FISH with newly designed probes for both groups specifically stained filamentous bacteria within the mats. FISH-based quantification of the two filament groups in six different microbial mat samples from Lower Kane Cave showed that LKC group II dominated five of the six mat communities. This study further expands our perceptions of the diversity and geographic distribution of "Epsilonproteobacteria" in extreme environments and demonstrates their biogeochemical importance in subterranean ecosystems.
Anaerobic ammonium oxidation by marine and freshwater planctomycete-like bacteria
2003 - Appl. Microbiol. Biotech., 63: 107–114
Abstract:
Recently, two fresh water species, " Candidatus Brocadia anammoxidans" and " Candidatus Kuenenia stuttgartiensis", and one marine species, " Candidatus Scalindua sorokinii", of planctomycete anammox bacteria have been identified. " Candidatus Scalindua sorokinii" was discovered in the Black Sea, and contributed substantially to the loss of fixed nitrogen. All three species contain a unique organelle--the anammoxosome--in their cytoplasm. The anammoxosome contains the hydrazine/hydroxylamine oxidoreductase enzyme, and is thus the site of anammox catabolism. The anammoxosome is surrounded by a very dense membrane composed almost exclusively of linearly concatenated cyclobutane-containing lipids. These so-called 'ladderanes' are connected to the glycerol moiety via both ester and ether bonds. In natural and man-made ecosystems, anammox bacteria can cooperate with aerobic ammonium-oxidising bacteria, which protect them from harmful oxygen, and provide the necessary nitrite. The cooperation of these two groups of ammonium-oxidising bacteria is the microbial basis for a sustainable one reactor system, CANON (completely autotrophic nitrogen-removal over nitrite) to remove ammonia from high strength wastewater.
Fluorescence in situ hybridisation for the identification and charcterisation of prokaryotes
2003 - Curr. Opin. Microbiol., 6: 302-309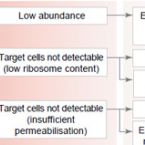
Abstract:
Fluorescence in situ hybridisation with rRNA-targeted nucleic acid probes can be used to directly identify microorganisms within complex samples in a few hours and therefore has widespread application in environmental and medical microbiology. The past year has seen significant methodological improvements in fluorescence in situ hybridisation, as well as in the combination of this method with other techniques for inferring functional traits of microorganisms within their environment.
Substrate uptake in extremely halophilic microbial communities revealed by microautoradiography and fluorescence in situ hybridization
2003 - Extremophiles, 7: 409-413
Abstract:
The combination of fluorescence in situ hybridization and microautoradiography (FISH-MAR approach) was applied to brine samples of a solar saltern crystallizer pond from Mallorca (Spain) where the simultaneous occurrence of Salinibacter spp. and the conspicuous square Archaea had been detected. Radioactively labeled bicarbonate, acetate, glycerol, and an amino acid mixture were tested as substrates for the microbial populations inhabiting such brines. The results indicated that hitherto uncultured 'square Archaea' do actively incorporate amino acids and acetate. However, Salinibacter spp. only showed amino acid incorporation in pure culture, but no evidence of such activity in their natural environment could be demonstrated. No glycerol incorporation was observed for any component of the microbial community.
Community analysis of ammonia and nitrite oxidizers during start-up of nitritation reactors
2003 - Appl. Environ. Microbiol., 69: 3213-3222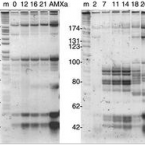
Abstract:
Partial nitrification of ammonium to nitrite under oxic conditions (nitritation) is a critical process for the effective use of alternative nitrogen removal technologies from wastewater. Here we investigated the conditions which promote establishment of a suitable microbial community for performing nitritation when starting from regular sewage sludge. Reactors were operated in duplicate under different conditions (pH, temperature, and dilution rate) and were fed with 50 mM ammonium either as synthetic medium or as sludge digester supernatant. In all cases, stable nitritation could be achieved within 10 to 20 days after inoculation. Quantitative in situ hybridization analysis with group-specific fluorescent rRNA-targeted oligonucleotides (FISH) in the different reactors showed that nitrite-oxidizing bacteria of the genus Nitrospira were only active directly after inoculation with sewage sludge (up to 4 days and detectable up to 10 days). As demonstrated by quantitative FISH and restriction fragment length polymorphism (RFLP) analyses of the amoA gene (encoding the active-site subunit of the ammonium monooxygenase), the community of ammonia-oxidizing bacteria changed within the first 15 to 20 days from a more diverse set of populations consisting of members of the Nitrosomonas communis and Nitrosomonas oligotropha sublineages and the Nitrosomonas europaea-Nitrosomonas eutropha subgroup in the inoculated sludge to a smaller subset in the reactors. Reactors operated at 30 degrees C and pH 7.5 contained reproducibly homogeneous communities dominated by one amoA RFLP type from the N. europaea-N. eutropha group. Duplicate reactors at pH 7.0 developed into diverse communities and showed transient population changes even within the ammonia oxidizer community. Reactors at pH 7.5 and 25 degrees C formed communities that were indistinguishable by the applied FISH probes but differing in amoA RFLP types. Communities in reactors fed with sludge digester supernatant exhibited a higher diversity and were constantly reinoculated with ammonium oxidizers from the supernatant. Therefore, such systems could be maintained at a higher dilution rate (0.75 day(-1) compared to 0.2 day(-1) for the synthetic wastewater reactors). Despite similar reactor performance with respect to chemical parameters, the underlying community structures were different, which may have an influence on stability during perturbations.
Characterization of activated sludge flocs by confocal laser scanning microscopy and image analysis
2003 - Wat. Res., 37: 2043–2052
Abstract:
In this study we present a new approach to determine volumes, heterogeneity factors, and compositions of the bacterial population of activated sludge flocs by 3D confocal imaging. After staining the fresh flocs with fluorescein-isothiocyanate, 75 stacks of images (containing approx. 3000 flocs) were acquired with a confocal laser scanning microscope. The self-developed macro 3D volume and surface determination for the KS 400 software package combined the images of one stack to a 3D image and calculated the real floc volume and surface. We determined heterogeneity factors like the ratio of real floc surface to the surface of a sphere with the respective volume and the fractal dimension (D(f)). According to their significant influence on floc integrity and quality, we also investigated the chemical composition of flocs and quantified their bacterial population structure by using group-specific rRNA-targeted probes for fluorescence in situ hybridization. By a settling experiment we enriched flocs with poor settling properties and determined the above-mentioned parameters. This approach revealed shifts in floc volume, heterogeneity, and bacterial and chemical composition according to the settling quality of the flocs.
Related assemblages of sulphate-reducing bacteria associated with ultradeep gold mines of South Africa and deep basalt aquifers of Washington State
2003 - Environ. Microbiol., 5: 267-277Abstract:
We characterized the diversity of sulphate-reducing bacteria (SRB) associated with South African gold mine boreholes and deep aquifer systems in Washington State, USA. Sterile cartridges filled with crushed country rock were installed on two hydrologically isolated and chemically distinct sites at depths of 3.2 and 2.7 km below the land surface (kmbls) to allow development of biofilms. Enrichments of sulphate-reducing chemolithotrophic (H2) and organotrophic (lactate) bacteria were established from each site under both meso- and thermophilic conditions. Dissimilatory sulphite reductase (Dsr) and 16S ribosomal RNA (rRNA) genes amplified from DNA extracted from the cartridges were most closely related to the Gram-positive species Desulfotomaculum thermosapovorans and Desulfotomaculum geothermicum, or affiliated with a novel deeply branching clade. The dsr sequences recovered from the Washington State deep aquifer systems affiliated closely with the South African sequences, suggesting that Gram-positive sulphate-reducing bacteria are widely distributed in the deep subsurface.
Nucleic acid-based, cultivation-independent detection of Listeria spp. and genotypes of L. monocytogenes
2003 - FEMS Immunol. Med. Microbiol., 35: 215-225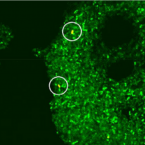
Abstract:
Based on comparative analysis of 16S rRNA gene sequences, two oligonucleotide probes for in situ detection of all members of the genus Listeria were designed. These probes allowed fast and reliable in situ detection of Listeria spp. even in complex samples like raw milk. Almost full-length iap (invasion-associated protein) gene sequences were determined for 69 Listeria monocytogenes strains of all 13 known serotypes. A comparison of these sequences revealed that the L. monocytogenes strains can be grouped into three distinct genotypes. These clusters correlate well with distinct serotypes. Thus, strains of serotypes b and d belong to genotype I, a and c to genotype II, and 4a and 4c, which are rarely isolated from humans, group together within genotype III. These results could be corroborated by further comparative sequence analysis of genes encoding two phospholipases - plcA and plcB. Based on the iap gene sequences, a highly specific and reproducible competitive PCR detection method was developed. Primer pairs targeting genotype-specific regions of the iap gene were designed. The amplification of non-specific PCR products from DNA of non-target strains was prevented by adding competitive primers. By applying this method, the rapid and reliable distinction of the three L. monocytogenes genotypes was possible.
probeBase - an online resource for rRNA-targeted oligonucleotide probes
2003 - Nucleic Acids Res., 31: 514-516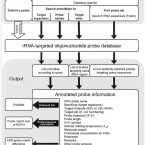
Abstract:
Ribosomal RNA-(rRNA)-targeted oligonucleotide probes are widely used for culture-independent identification of microorganisms in environmental and clinical samples. ProbeBase is a comprehensive database containing more than 700 published rRNA-targeted oligonucleotide probe sequences (status August 2002) with supporting bibliographic and biological annotation that can be accessed through the internet at http://www.probebase.net. Each oligonucleotide probe entry contains information on target organisms, target molecule (small- or large-subunit rRNA) and position, G+C content, predicted melting temperature, molecular weight, necessity of competitor probes, and the reference that originally described the oligonucleotide probe, including a link to the respective abstract at PubMed. In addition, probes successfully used for fluorescence in situ hybridization (FISH) are highlighted and the recommended hybridization conditions are listed. ProbeBase also offers difference alignments for 16S rRNA-targeted probes by using the probe match tool of the ARB software and the latest small-subunit rRNA ARB database (release June 2002). The option to directly submit probe sequences to the probe match tool of the Ribosomal Database Project II (RDP-II) further allows one to extract supplementary information on probe specificities. The two main features of probeBase, 'search probeBase' and 'find probe set', help researchers to find suitable, published oligonucleotide probes for microorganisms of interest or for rRNA gene sequences submitted by the user. Furthermore, the 'search target site' option provides guidance for the development of new FISH probes.
Modern scientific methods and their potential in wastewater science and technology
2002 - Water Res., 36: 370-93
Abstract:
Application of novel analytical and investigative methods such as fluorescence in situ hybridization, confocal laser scanning microscopy (CLSM), microelectrodes and advanced numerical simulation has led to new insights into micro- and macroscopic processes in bioreactors. However, the question is still open whether or not these new findings and the subsequent gain of knowledge are of significant practical relevance and if so, where and how. To find suitable answers it is necessary for engineers to know what can be expected by applying these modern analytical tools. Similarly, scientists could benefit significantly from an intensive dialogue with engineers in order to find out about practical problems and conditions existing in wastewater treatment systems. In this paper, an attempt is made to help bridge the gap between science and engineering in biological wastewater treatment. We provide an overview of recently developed methods in microbiology and in mathematical modeling and numerical simulation. A questionnaire is presented which may help generate a platform from which further technical and scientific developments can be accomplished. Both the paper and the questionnaire are aimed at encouraging scientists and engineers to enter into an intensive, mutually beneficial dialogue.
The anammox case - A new experimental manifesto for microbiological eco-physiology
2002 - Antonie van Leeuwenhoek, 81: 693-702The genus Caedibacter comprises endosymbionts of Paramecium spp. related to the Rickettsiales (Alphaproteobacteria) and to Francisella tularensis (Gammaproteobacteria)
2002 - Appl. Environ. Microbiol., 68: 6043-6050
Abstract:
Obligate bacterial endosymbionts of paramecia able to form refractile inclusion bodies (R bodies), thereby conferring a killer trait upon their ciliate hosts, have traditionally been grouped into the genus CAEDIBACTER: Of the six species described to date, only the Paramecium caudatum symbiont Caedibacter caryophilus has been phylogenetically characterized by its 16S rRNA gene sequence, and it was found to be a member of the Alphaproteobacteria related to the RICKETTSIALES: In this study, the Caedibacter taeniospiralis type strain, an R-body-producing cytoplasmatic symbiont of Paramecium tetraurelia strain 51k, was investigated by comparative 16S rRNA sequence analysis and fluorescence in situ hybridization with specific oligonucleotide probes. C. taeniospiralis is not closely related to C. caryophilus (80% 16S rRNA sequence similarity) but forms a novel evolutionary lineage within the Gammaproteobacteria with the family Francisellaceae as a sister group (87% 16S rRNA sequence similarity). These findings demonstrate that the genus Caedibacter is polyphyletic and comprises at least two phylogenetically different bacterial species belonging to two different classes of the PROTEOBACTERIA: Comparative phylogenetic analysis of C. caryophilus, five closely related Acanthamoeba endosymbionts (including one previously uncharacterized amoebal symbiont identified in this study), and their hosts suggests that the progenitor of the alphaproteobacterial C. caryophilus lived within acanthamoebae prior to the infection of paramecia.
Microbial community composition and function in wastewater treatment plants
2002 - Antonie van Leeuwenhoek, 81: 665-680
Abstract:
Biological wastewater treatment has been applied for more than a century to ameliorate anthropogenic damage to the environment. But only during the last decade the use of molecular tools allowed to accurately determine the composition, and dynamics of activated sludge and biofilm microbial communities. Novel, in many cases yet not cultured bacteria were identified to be responsible for filamentous bulking and foaming as well as phosphorus and nitrogen removal in these systems. Now, methods are developed to infer the in situ physiology of these bacteria. Here we provide an overview of what is currently known about the identity and physiology of some of the microbial key players in activated sludge and biofilm systems.
Origins and diversification of sulfate-respiring microorganisms
2002 - Antonie van Leeuwenhoek, 81: 189-195Abstract:
If the diversification of microbial life can be depicted as a single tree, as inferred by comparative sequencing of ribosomal RNAs, this could provide a framework for defining the order of emergence of new metabolic pathways. However, recent recognition that lateral gene transfer has been a significant force in microbial evolution has created uncertainty about the interpretation of taxonomies based on gene sequences. In this context, the origins and evolution of sulfate respiration will be evaluated considering the evolutionary history of a central enzyme in this process, the dissimilatory sulfite reductase. These studies suggest at least two major lateral transfer events during the early diversification of sulfate respiring microorganisms. The high sequence conservation of this enzyme has also provided a mechanism to directly explore the natural diversity of sulfate-respiring organisms using molecular techniques, avoiding the bias of culture-based identification. These studies suggest that the habitat range and evolutionary diversity of this key functional group of organisms is greater than now appreciated.
Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment
2002 - Appl. Environ. Microbiol., 68: 5064-5081
Abstract:
For cultivation-independent detection of sulfate-reducing prokaryotes (SRPs) an oligonucleotide microarray consisting of 132 16S rRNA gene-targeted oligonucleotide probes (18-mers) having hierarchical and parallel (identical) specificity for the detection of all known lineages of sulfate-reducing prokaryotes (SRP-PhyloChip) was designed and subsequently evaluated with 41 suitable pure cultures of SRPs. The applicability of SRP-PhyloChip for diversity screening of SRPs in environmental and clinical samples was tested by using samples from periodontal tooth pockets and from the chemocline of a hypersaline cyanobacterial mat from Solar Lake (Sinai, Egypt). Consistent with previous studies, SRP-PhyloChip indicated the occurrence of Desulfomicrobium spp. in the tooth pockets and the presence of Desulfonema- and Desulfomonile-like SRPs (together with other SRPs) in the chemocline of the mat. The SRP-PhyloChip results were confirmed by several DNA microarray-independent techniques, including specific PCR amplification, cloning, and sequencing of SRP 16S rRNA genes and the genes encoding the dissimilatory (bi)sulfite reductase (dsrAB).
Deammonification in biofilm systems: Population structure and function
2002 - Wat. Sci. Tech., 46: 223-231Abstract:
For the development of alternative concepts for the cost effective treatment of wastewaters with high ammonium content and low C/N-ratio, autotrophic consortia of micro-organisms with the ability to convert ammonium directly into N2 are of particular interest. Several full-scale industrial biofilm plants eliminating nitrogen without carbon source for years in a stable process, are suspected for some time to harbor active anaerobic ammonium oxidizers in deeper, oxygen-limited biofilm layers. In order to identify the processes of the single-stage nitrogen elimination (deammonification) in biofilm systems and to allocate them to the responsible micro-organisms, a deammonifying moving-bed pilot plant was investigated in detail. 15N-labelled tracer compounds were used as well as 16S rDNA libraries and in situ identification of dominant organisms. The usage of rRNA-targeted oligonucleotide probes (FISH) was particularly emphasized on the ammonium oxidizers of the beta-subclass of Proteobacteria and on the members of the order Planctomycetales. The combined application of these methods led to a deeper insight into the population structure and function of a deammonifying biofilm.
Population dynamics in wastewater treatment plants with enhanced biological phosphorous removal operated with and without nitrogen removal
2002 - Wat. Sci. Tech., 46: 163-170Abundance and phylogenetic affiliation of iron-reducers in activated sludge as assessed by fluorescence in situ hybridization and microautoradiography
2002 - Appl. Environ. Microbiol., 68: 4629-4636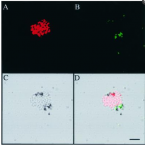
Abstract:
Microautoradiography (MAR) was used to enumerate acetate-consuming bacteria under Fe(III)-reducing conditions in activated sludge. This population is believed to consist of dissimilatory iron-reducing bacteria, because the applied incubation conditions and the use of specific inhibitors excluded consumption of radiolabeled acetate by other physiological groups such as sulfate reducers. By use of this approach, dissimilatory iron reducers were found in a concentration of 1.1 x 10(8) cells per ml, corresponding to approximately 3% of the total cell count as determined by DAPI (4',6'-diamino-2-phenylindoledihydrochloride-dilactate) staining. The MAR enumeration method was compared to the traditional most-probable-number (MPN) method (FeOOH-MPN) and a modified MPN method, which contains Ferrozine directly within the MPN dilutions to determine the production of small amounts of ferrous iron (Ferrozine-MPN). The Ferrozine-MPN method yielded values 6 to 10 times higher than those obtained by the FeOOH-MPN method. Nevertheless, the MAR approach yielded counts that were 100 to 1,000 times higher than those obtained by the Ferrozine-MPN method. Specific in situ Fe(III) reduction rates per cell (enumerated by the MAR method) were calculated and found to be comparable to the respective rates for pure cultures of dissimilatory iron-reducing bacteria, suggesting that the new MAR method is most reliable. A combination of MAR and fluorescence in situ hybridization was used for phylogenetic characterization of the putative iron-reducing bacteria. All activated-sludge cells able to consume acetate under iron-reducing conditions were targeted by the bacterial oligonucleotide probe EUB338. Around 20% were identified as gamma Proteobacteria, and 10% were assigned to the delta subclass of Proteobacteria.
Molecular evidence for a uniform microbial community in sponges from different oceans
2002 - Appl. Environ. Microbiol., 68: 4431-4440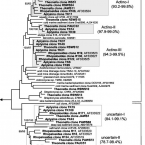
Abstract:
Sponges (class Porifera) are evolutionarily ancient metazoans that populate the tropical oceans in great abundances but also occur in temperate regions and even in freshwater. Sponges contain large numbers of bacteria that are embedded within the animal matrix. The phylogeny of these bacteria and the evolutionary age of the interaction are virtually unknown. In order to provide insights into the species richness of the microbial community of sponges, we performed a comprehensive diversity survey based on 190 sponge-derived 16S ribosomal DNA (rDNA) sequences. The sponges Aplysina aerophoba and Theonella swinhoei were chosen for construction of the bacterial 16S rDNA library because they are taxonomically distantly related and they populate nonoverlapping geographic regions. In both sponges, a uniform microbial community was discovered whose phylogenetic signature is distinctly different from that of marine plankton or marine sediments. Altogether 14 monophyletic, sponge-specific sequence clusters were identified that belong to at least seven different bacterial divisions. By definition, the sequences of each cluster are more closely related to each other than to a sequence from nonsponge sources. These monophyletic clusters comprise 70% of all publicly available sponge-derived 16S rDNA sequences, reflecting the generality of the observed phenomenon. This shared microbial fraction represents the smallest common denominator of the sponges investigated in this study. Bacteria that are exclusively found in certain host species or that occur only transiently would have been missed. A picture emerges where sponges can be viewed as highly concentrated reservoirs of so far uncultured and elusive marine microorganisms.
Bacterial community composition and function in sewage treatment systems
2002 - Curr. Opin. Biotechnol., 13: 218-227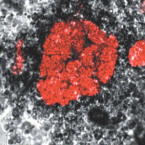
Abstract:
The application of modern molecular techniques has led to the identification, in situ quantification, and partial ecophysiological characterisation of bacteria responsible for bulking and foaming or for nutrient removal in sewage treatment systems. Unexpectedly, previously unrecognised, yet uncultured bacteria were demonstrated to catalyse nitrogen and phosphorous removal in activated-sludge and biofilm reactors. These findings provide the basis for the development of novel concepts for improving the efficiency and functional stability of waste water treatment systems.
Detection and differentiation of chlamydiae by fluorescence in situ hybridization (FISH)
2002 - Appl. Environ. Microbiol., 68: 4081-4089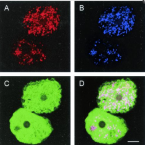
Abstract:
Chlamydiae are important pathogens of humans and animals but diagnosis of chlamydial infections is still hampered by inadequate detection methods. Fluorescence in situ hybridization (FISH) using rRNA-targeted oligonucleotide probes is widely used for the investigation of uncultured bacteria in complex microbial communities and has recently also been shown to be a valuable tool for the rapid detection of various bacterial pathogens in clinical specimens. Here we report on the development and evaluation of a hierarchic probe set for the specific detection and differentiation of chlamydiae, particularly C. pneumoniae, C. trachomatis, C. psittaci, and the recently described chlamydia-like bacteria comprising the novel genera Neochlamydia and PARACHLAMYDIA: The specificity of the nine newly developed probes was successfully demonstrated by in situ hybridization of experimentally infected amoebae and HeLa 229 cells, including HeLa 229 cells coinfected with C. pneumoniae and C. trachomatis. FISH reliably stained chlamydial inclusions as early as 12 h postinfection. The sensitivity of FISH was further confirmed by combination with direct fluorescence antibody staining. In contrast to previously established detection methods for chlamydiae, FISH was not susceptible to false-positive results and allows the detection of all recognized chlamydiae in one single step.
The microbial community composition of a nitrifying-denitrifying activated sludge from an industrial sewage treatment plant analyzed by the full-cycle rRNA approach
2002 - Syst. Appl. Microbiol., 25: 84-99
Abstract:
The composition of the microbial community present in the nitrifying-denitrifying activated sludge of an industrial wastewater treatment plant connected to a rendering facility was investigated by the full-cycle rRNA approach. After DNA extraction using three different methods, 94 almost full-length 16S rRNA gene clones were retrieved and analyzed phylogenetically. 59% of the clones were affiliated with the Proteobacteria and clustered with the beta- (29 clones), alpha- (24), and delta-class (2 clones), respectively. 15 clones grouped within the green nonsulfur (GNS) bacteria and 11 clones belonged to the Planctomycetes. The Verrucomicrobia, Acidobacteria, Nitrospira, Bacteroidetes, Firmicutes and Actinobacteria were each represented by one to five clones. Interestingly, the highest 'species richness' [measured as number of operational taxonomic units (OTUs)] was found within the alpha-class of Proteobacteria, followed by the Planctomycetes, the beta-class of Proteobacteria, and the GNS-bacteria. The microbial community composition of the activated sludge was determined quantitatively by using 36 group-, subgroup-, and OTU-specific rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization (FISH), confocal laser scanning microscopy and digital image analysis. 89% of all bacteria detectable by FISH with a bacterial probe set could be assigned to specific divisions. Consistent with the 16S rRNA gene library data, members of the beta-class of Proteobacteria dominated the microbial community and represented almost half of the biovolume of all bacteria detectable by FISH. Within the beta-class, 98% of the cells could be identified by the application of genus- or OTU-specific probes demonstrating a high in situ abundance of bacteria related to Zoogloea and Azoarcus sensu lato. Taken together, this study provides the first encompassing, high-resolution insight into the in situ composition of the microbial community present in a full-scale, industrial wastewater treatment plant.
Various bacterial pathogens and symbionts infect the amoeba Dictyostelium discoideum
2002 - Int. J. Med. Microbiol., 291: 615-624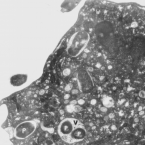
Abstract:
The haploid soil amoeba Dictyostelium discoideum is a suitable model organism to study host-pathogen interactions with Legionella pneumophila. In this study we show that D. discoideum AX2 is also susceptible to infection with other important human pathogens and obligate intracellular symbionts. Infection assays demonstrated that Legionella-like amoebal pathogens (LLAP K62), Mycobacterium avium and the obligate intracellular endosymbionts of Acanthamoeba sp. strains TUME1, UWE25 and UWC6 were able to multiply within Dictyostelium. Salmonella typhimurium and Pseudomonas aeruginosa also invaded Dictyostelium, however were degraded shortly after uptake. Comitin-minus host cells were more permissive to infections with L. pneumophila and LLAP K62. Furthermore, this mutation significantly delayed the degradation of S. typhimurium. Accompanying electron and fluorescence microscopy of infected AX2 cells revealed that L. pneumophila and M. avium replicate within vacuoles, while LLAP K62, TUME1 and UWE25 were tightly enclosed by membranous structures within the cytoplasm. The beta-proteobacterium UWC6 was found to persist in the cytoplasm. The observed subcellular locations which correspond to the locations within the respective natural hosts suggest that D. discoideum is a representative model system for these pathogens and symbionts.
Obligate bacterial endosymbionts of Acanthamoeba spp. related to the ß-subclass of Proteobacteria: proposal of 'Candidatus Procabacter acanthamoebae' gen. nov., sp. nov
2002 - Int. J. Syst. Evol. Microbiol., 52: 599–605
Abstract:
All obligate bacterial endosymbionts of free-living amoebae currently described are affiliated with the alpha-Proteobacteria, the Chlamydiales or the phylum Cytophaga-Flavobacterium-Bacteroides. Here, six rod-shaped gram-negative obligate bacterial endosymbionts of clinical and environmental isolates of Acanthamoeba spp. from the USA and Malaysia are reported. Comparative 16S rDNA sequence analysis demonstrated that these endosymbionts form a novel, monophyletic lineage within the beta-Proteobacteria, showing less than 90% sequence similarity to all other recognized members of this subclass. 23S rDNA sequence analysis of two symbionts confirmed this affiliation and revealed the presence of uncommon putative intervening sequences of 146 bp within helix-25 that shared no sequence homology to any other bacterial rDNA. In addition, the 23S rRNA of these endosymbionts displayed one polymorphism at the target site of oligonucleotide probe BET42a that is conserved in all other sequenced beta-Proteobacteria. Intra-cytoplasmatic localization of the endosymbionts within the amoebal host cells was confirmed by electron microscopy and fluorescence in situ hybridization with a specific 16S rRNA-targeted oligonucleotide probe. Based on these findings, the provisional name 'Candidatus Procabacter acanthamoebae' is proposed for classification of a representative of the six endosymbionts of Acanthamoeba spp. studied in this report. Comparative 18S rDNA sequence analysis of the Acanthamoeba host cells revealed their membership with either Acanthamoeba 18S rDNA sequence type T5 (Acanthamoeba lenticulata) or sequence type T4, which comprises the majority of all Acanthamoeba isolates.
Nitrifying and heterotrophic population dynamics in biofilm reactors: effects of hydraulic retention time and the presence of organic carbon
2002 - Wat. Res., 36: 469-481
Abstract:
Two biofilm reactors operated with hydraulic retention times of 0.8 and 5.0 h were used to study the links between population dynamics and reactor operation performance during a shift in process operation from pure nitrification to combined nitrification and organic carbon removal. The ammonium and the organic carbon loads were identical for both reactors. The composition and dynamics of the microbial consortia were quantified by fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes combined with confocal laser scanning microscopy, and digital image analysis. In contrast to past research, after addition of acetate as organic carbon nitrification performance decreased more drastically in the reactor with longer hydraulic retention time. FISH analysis showed that this effect was caused by the unexpected formation of a heterotrophic microorganism layer on top of the nitrifying biofilm that limited nitrifiers oxygen supply. Our results demonstrate that extension of the hydraulic retention time might be insufficient to improve combined nitrification and organic carbon removal in biofilm reactors.
16S-23S rDNA intergenic spacer and 23S rDNA of anaerobic ammonium oxidizers: implications for phylogeny and in situ detection
2001 - Environ. Microbiol., 3: 450-459Nitrification performance and nitrifier community composition of a chemostat and a membrane-assisted bioreactor for the nitrification of sludge reject waters
2001 - Bioprocess. Biosyst. Eng., 24: 203-210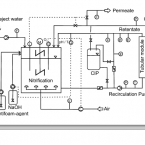
Microbiology and application of the anoxic ammonium oxidation 'Anammox' process
2001 - Curr. Opin. Biotechnol., 12: 283-288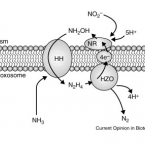
Quantification of bacterial populations in complex ecosystems using fluorescent in situ hybridization, confocal laser scanning microscopy and image analysis
2001 - Genetics Selection Evolution, 33: S307-S318Cultivation-independent, semiautomatic determination of absolute bacterial cell numbers in environmental samples by fluorescence in situ hybridization
2001 - Appl. Environ. Microbiol., 67: 5810-5818
Abstract:
Fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes has found widespread application for analyzing the composition of microbial communities in complex environmental samples. Although bacteria can quickly be detected by FISH, a reliable method to determine absolute numbers of FISH-stained cells in aggregates or biofilms has, to our knowledge, never been published. In this study we developed a semiautomated protocol to measure the concentration of bacteria (in cells per volume) in environmental samples by a combination of FISH, confocal laser scanning microscopy, and digital image analysis. The quantification is based on an internal standard, which is introduced by spiking the samples with known amounts of Escherichia coli cells. This method was initially tested with artificial mixtures of bacterial cultures and subsequently used to determine the concentration of ammonia-oxidizing bacteria in a municipal nitrifying activated sludge. The total number of ammonia oxidizers was found to be 9.8 x 10(7) +/- 1.9 x 10(7) cells ml(-1). Based on this value, the average in situ activity was calculated to be 2.3 fmol of ammonia converted to nitrite per ammonia oxidizer cell per h. This activity is within the previously determined range of activities measured with ammonia oxidizer pure cultures, demonstrating the utility of this quantification method for enumerating bacteria in samples in which cells are not homogeneously distributed.
Evidence for additional genus-level diversity of Chlamydiales in the environment
2001 - FEMS Microbiol. Lett., 204: 71-74
Abstract:
The medically important order Chlamydiales has long been considered to contain a few closely related bacteria which occur exclusively in animals and humans. This perception of diversity and habitat had to be revised with the recent identification of the genera Simkania, Waddlia, Parachlamydia, and Neochlamydia with the latter two comprising endosymbionts of amoebae. Application of a newly developed PCR assay for the specific amplification of a near full length 16S rDNA fragment of these novel Chlamydia-related bacteria on activated sludge samples revealed the existence of at least four additional, previously unknown evolutionary lineages of Chlamydiales (each showing less than 92% 16S rRNA sequence similarity with all recognized members of this order). These findings suggest that some waste water treatment plants represent reservoirs for a diverse assemblage of environmental chlamydiae, a discovery which might also be of relevance from the viewpoint of human public health.
In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in waste water treatment plants
2001 - Appl. Environ. Microbiol., 67: 5273-5284
Abstract:
Uncultivated Nitrospira-like bacteria in different biofilm and activated-sludge samples were investigated by cultivation-independent molecular approaches. Initially, the phylogenetic affiliation of Nitrospira-like bacteria in a nitrifying biofilm was determined by 16S rRNA gene sequence analysis. Subsequently, a phylogenetic consensus tree of the Nitrospira phylum including all publicly available sequences was constructed. This analysis revealed that the genus Nitrospira consists of at least four distinct sublineages. Based on these data, two 16S rRNA-directed oligonucleotide probes specific for the phylum and genus Nitrospira, respectively, were developed and evaluated for suitability for fluorescence in situ hybridization (FISH). The probes were used to investigate the in situ architecture of cell aggregates of Nitrospira-like nitrite oxidizers in wastewater treatment plants by FISH, confocal laser scanning microscopy, and computer-aided three-dimensional visualization. Cavities and a network of cell-free channels inside the Nitrospira microcolonies were detected that were water permeable, as demonstrated by fluorescein staining. The uptake of different carbon sources by Nitrospira-like bacteria within their natural habitat under different incubation conditions was studied by combined FISH and microautoradiography. Under aerobic conditions, the Nitrospira-like bacteria in bioreactor samples took up inorganic carbon (as HCO(3)(-) or as CO(2)) and pyruvate but not acetate, butyrate, and propionate, suggesting that these bacteria can grow mixotrophically in the presence of pyruvate. In contrast, no uptake by the Nitrospira-like bacteria of any of the carbon sources tested was observed under anoxic or anaerobic conditions.
Multiple lateral transfer events of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes
2001 - J. Bacteriol., 183: 6028-6035
Members of the Cytophaga-Flavobacterium-Bacteroides phylum as intracellular bacteria of acanthamoebae: proposal of 'Candidatus Amoebophilus asiaticus'
2001 - Environ. Microbiol., 3: 440-449
Abstract:
Three Gram-negative, rod-shaped bacteria that were found intracellularly in two environmental and one clinical Acanthamoeba sp. isolates were analysed. Two endocytobionts showing a parasitic behaviour were propagated successfully outside their amoebal host cells and were identified subsequently by comparative 16S rRNA sequence analysis as being most closely affiliated with Flavobacterium succinicans (99% 16S rRNA sequence similarity) or Flavobacterium johnsoniae (98% 16S rRNA sequence similarity). One endocytobiont could neither be cultivated outside its original Acanthamoeba host (Acanthamoeba sp. TUMSJ-321) nor transferred into other amoebae. Electron microscopy revealed that the amoebal trophozoites and cysts were almost completely filled with cells of this endosymbiont which are surrounded by a host-derived membrane. According to 16S rRNA sequence analysis, this endosymbiont could also be assigned to the Cytophaga-Flavobacterium-Bacteroides (CFB) phylum, but was not closely affiliated to any recognized species within this phylogenetic group (less than 82% 16S rRNA sequence similarity). Identity and intracellular localization of this endosymbiont were confirmed by application of a specific fluorescently labelled 16S rRNA-targeted probe. Based on these findings, we propose classification of this obligate Acanthamoeba endosymbiont as 'Candidatus Amoebophilus asiaticus'. Comparative 18S rRNA sequence analysis of the host of 'Candidatus Amoebophilus asiaticus' revealed its membership with Acanthamoeba 18S rDNA sequence type T4 that comprises the majority of all Acanthamoeba isolates.
Isolation and properties of obligately chemolithoautotrophic and extremely alkalitolerant ammonia oxidizing bacteria from Mongolian soda lakes
2001 - Arch. Microbiol., 176: 170-177
Abstract:
Five mixed samples prepared from the surface sediments of 20 north-east Mongolian soda lakes with total salt contents from 5 to 360 g/l and pH values from 9.7 to 10.5 were used to enrich for alkaliphilic ammonia-oxidizing bacteria. Successful enrichments at pH 10 were achieved on carbonate mineral medium containing 0.6 M total Na(+) and < or =4 mM NH(4)Cl. Five isolates (ANs1-ANs5) of ammonia-oxidizing bacteria capable of growth at pH 10 were obtained from the colonies developed on bilayered gradient plates. The cells were motile and coccoid, with well-developed intracytoplasmic membranes (ICPM) and carboxysomes. At pH 10.0, ammonia was toxic for growth at concentrations higher than 5 mM NH(4)Cl. The bacteria were able to grow within the salinity range of 0.1-1.0 M of total Na+ (optimum 0.3 M). In media containing 0.3-0.6 M total Na(+), optimal growth in batch cultures occurred in the presence of a bicarbonate/carbonate buffer system within the pH range 8.5-9.5, with the highest pH limit at pH 10.5. At pH lower than 8.0, growth was slower, most probably due to decreasing free ammonia. The pH profile of the respiratory activity was broader, with limits at 6.5-7.0 and 11.0 and an optimum at 9.5-10.0. In pH-controlled, NH(3)-limited continuous culture, isolate ANs5 grew up to pH 11.3, which is the highest pH limit known for ammonia-oxidizing bacteria so far. This showed the existence of extremely alkali-tolerant ammonia-oxidizing bacteria in the soda lakes. Comparative 16S rDNA sequence analysis of the five isolates demonstrated that they possess identical 16S rDNA genes and that they are closely related to Nitrosomonas halophila (sequence similarity 99.3%), a member of the beta-subclass of the Proteobacteria. This affiliation was confirmed by comparative sequence analysis of the amoA gene, encoding the active-site subunit of the ammonia-monoxygenase, of one of the isolates. DNA-DNA hybridization data further supported that the soda lake isolates are very similar to each other and represent an alkali-tolerant subpopulation of N. halophila whose species description is herewith amended.
Assessment of metabolic potential of biofilm-associated bacteria
2001 - Methods Enzymol., 336: 265-76
Nitrification in sequencing biofilm batch reactors: lessons from molecular approaches
2001 - Water Sci. Technol., 43: 9-18
Abstract:
The nitrifying microbial diversity and population structure of a sequencing biofilm batch reactor receiving sewage with high ammonia and salt concentrations (SBBR 1) was analyzed by the full-cycle rRNA approach. The diversity of ammonia-oxidizers in this reactor was additionally investigated using comparative sequence analysis of a gene fragment of the ammonia monooxygenase (amoA), which represents a key enzyme of all ammonia-oxidizers. Despite the "extreme" conditions in the reactor, a surprisingly high diversity of ammonia- and nitrite-oxidizers was observed to occur within the biofilm. In addition, molecular evidence for the existence of novel ammonia-oxidizers is presented. Quantification of ammonia- and nitrite-oxidizers in the biofilm by Fluorescent In situ Hybridization (FISH) and digital image analysis revealed that ammonia-oxidizers occurred in higher cell numbers and occupied a considerably larger share of the total biovolume than nitrite-oxidizing bacteria. In addition, ammonia oxidation rates per cell were calculated for different WWTPs following the quantification of ammonia-oxidizers by competitive PCR of an amoA gene fragment. The morphology of nitrite-oxidizing, unculturable Nitrospira-like bacteria was studied using FISH, confocal laser scanning microscopy (CLSM) and three-dimensional visualization. Thereby, a complex network of microchannels and cavities was detected within microcolonies of Nitrospira-like bacteria. Microautoradiography combined with FISH was applied to investigate the ability of these organisms to use CO2 as carbon source and to take up other organic substrates under varying conditions. Implications of the obtained results for fundamental understanding of the microbial ecology of nitrifiers as well as for future improvement of nutrient removal in wastewater treatment plants (WWTPs) are discussed.
Endosymbiotic sulphate-reducing and sulphide-oxidizing bacteria in an oligochaete worm
2001 - Nature, 411: 298-302
Isolation and phylogenetic analysis of bacteria with antimicrobial activities from the Mediterranean sponges Aplysina aerophoba and Aplysina cavernicola
2001 - FEMS Microbiol. Ecol., 35: 305-312
Abstract:
The aim of this study was to isolate bacteria with antimicrobial activities from the marine sponges Aplysina aerophoba and Aplysina cavernicola. The obtained 27 isolates could be subdivided into eight phylogenetically different clusters based on comparative sequence analysis of their 16S rDNA genes. The sponge isolates were affiliated with the low (Bacillus) and high G+C Gram-positive bacteria (Arthobacter, Micrococcus), as well as the alpha-Proteobacteria (unknown isolate) and gamma-Proteobacteria (Vibrio, Pseudoalteromonas). One novel Bacillus species was identified and two species were closely related to previously uncharacterized strains. Isolates with antimicrobial activity were numerically most abundant in the genera Pseudoalteromonas and the alpha-Proteobacteria. The sponge isolates show antimicrobial activities against Gram-positive and Gram-negative reference strains but not against the fungus Candida albicans. A general pattern was observed in that Gram-positive bacteria inhibited Gram-positive strains while Gram-negative bacteria inhibited Gram-negative isolates. Antimicrobial activities were also found against clinical isolates, i.e. multi-resistant Staphylococcus aureus and Staphylococcus epidermidis strains isolated from hospital patients. The high recovery of strains with antimicrobial activity suggests that marine sponges represent an ecological niche which harbors a hitherto largely uncharacterized microbial diversity and, concomitantly, a yet untapped metabolic potential.
Community structure and activity dynamics of nitrifying bacteria in a phosphate-removing biofilm
2001 - Appl. Environ. Microbiol., 67: 1351-1362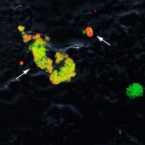
Abstract:
The microbial community structure and activity dynamics of a phosphate-removing biofilm from a sequencing batch biofilm reactor were investigated with special focus on the nitrifying community. O(2), NO(2)(-), and NO(3)(-) profiles in the biofilm were measured with microsensors at various times during the nonaerated-aerated reactor cycle. In the aeration period, nitrification was oxygen limited and restricted to the first 200 microm at the biofilm surface. Additionally, a delayed onset of nitrification after the start of the aeration was observed. Nitrate accumulating in the biofilm in this period was denitrified during the nonaeration period of the next reactor cycle. Fluorescence in situ hybridization (FISH) revealed three distinct ammonia-oxidizing populations, related to the Nitrosomonas europaea, Nitrosomonas oligotropha, and Nitrosomonas communis lineages. This was confirmed by analysis of the genes coding for 16S rRNA and for ammonia monooxygenase (amoA). Based upon these results, a new 16S rRNA-targeted oligonucleotide probe specific for the Nitrosomonas oligotropha lineage was designed. FISH analysis revealed that the first 100 microm at the biofilm surface was dominated by members of the N. europaea and the N. oligotropha lineages, with a minor fraction related to N. communis. In deeper biofilm layers, exclusively members of the N. oligotropha lineage were found. This separation in space and a potential separation of activities in time are suggested as mechanisms that allow coexistence of the different ammonia-oxidizing populations. Nitrite-oxidizing bacteria belonged exclusively to the genus Nitrospira and could be assigned to a 16S rRNA sequence cluster also found in other sequencing batch systems.
Quantification of biofilms in multi-spectral digital volumes from confocal laser-scanning microscopes
2000 - Image Anal. Stereol., 19: 151-156
Phylogeny of all recognized species of ammonia-oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys
2000 - Appl. Environ. Microbiol., 66: 5368-5382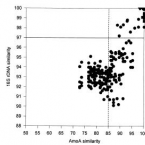
Novel Nitrospira-like bacteria as dominant nitrite-oxidizers in biofilms from wastewater treatment plants: Diversity and in situ physiology
2000 - Water Sci. Technol., 41: 85-90
Successfull and unsuccessfull bioaugmentation experiments monitored by fluorescent in situ hybridization
2000 - Wat. Sci. Tech., 41: 61-68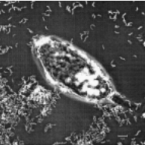
Effect of long-term idle periods on the performance of sequencing batch reactors
2000 - Wat. Sci. Tech., 41: 105-113
Ecological study of a bioaugmentation failure
2000 - Environ. Microbiol., 2: 179-190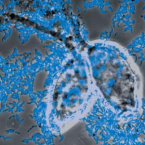
Abstract:
A nitrifying sequencing batch reactor was inoculated twice with the aerobic denitrifying bacterium Microvirgula aerodenitrificans and fed with acetate. No improvement was obtained on nitrogen removal. The second more massive inoculation was even followed by a nitrification breakdown, while at the same time, nitrification remained stable in a second reactor operated under the same conditions without bioaugmentation. Fluorescent in situ hybridization with rRNA-targeted probes revealed that the added bacteria almost disappeared from the reactor within 2 days, and that digestive vacuoles of protozoa gave strong hybridization signals with the M. aerodenitrificans-specific probe. An overgrowth of protozoa, coincident with the disappearance of free-living bacteria, was monitored by radioactive dot-blot hybridization only in the bioaugmented reactor. Population dynamics were analysed with a newly developed in situ quantification procedure of the probe-targeted bacteria. The nitrifying groups of bacteria decreased in a similar way in the bioaugmented and non-bioaugmented reactors. Other bacterial groups evolved differently. The involvement of different ecological parameters are discussed separately for each reactor. These results underline the importance of predator-prey interaction and illustrate the undesirable effects of massive bioaugmentation.
Phylogenetic relationships of filamentous bacteria of the Eikelboom Type 021N isolated from bulking activated sludge and development of an encompassing set of 021N-specific oligonucleotide probes
2000 - Appl. Environ. Microbiol., 66: 5043-5052
Abstract:
Fifteen filamentous strains, morphologically classified as Eikelboom type 021N bacteria, were isolated from bulking activated sludges. Based on comparative 16S ribosomal DNA (rDNA) sequence analysis, all strains form a monophyletic cluster together with all recognized Thiothrix species (88.3 to 98.7% 16S rDNA sequence similarity) within the gamma-subclass of Proteobacteria. The investigated Eikelboom type 021N isolates were subdivided into three distinct groups (I to III) demonstrating a previously unrecognized genetic diversity hidden behind the uniform morphology of the filaments. For in situ detection of these bacteria, 16S rRNA-targeted oligonucleotide probes specific for the entire Eikelboom type 021N-Thiothrix cluster and the Eikelboom type 021N groups I, II, and III, respectively, were designed, evaluated, and successfully applied in activated sludge.
Molecular evidence for genus level diversity of bacteria capable of catalyzing anaerobic ammonium oxidation
2000 - Syst. Appl. Microbiol, 23: 93-106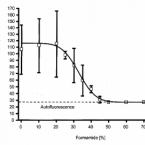
Abstract:
Recently, a bacterium capable to oxidize ammonium anaerobically at a high rate was identified as novel member of the Planctomycetales (Strous, M., Fuersi, J. A., Kramer, E. H. M., Logemann, S., Muyzer, G., van de Pas-Schoonen, K. T., Webb, R. I., Kufnen, J. G., and Jetten, M. S. M.: Nature 400, 446-449, 1999). Here we investigated the microbial community structure of a trickling filter biofilm with a high anaerobic ammonium oxidation activity. Fluorescence in situ hybridization (FISH) with a set of nine probes designed for specific identification of the recently described anaerobic ammonium oxidizer demonstrated that only one probe hybridized to bacteria within the biofilm. For phylogenetic characterization of putative biofilm anaerobic ammonium oxidizers a full-cycle 16S rDNA approach was performed by using a Planctomycetales-specific forward primer for PCR amplification. Of the twenty-five 16S rDNA fragments (1364 bp in length) amplified from the biofilm, nine were affiliated to the Planctomycetales. Comparative analysis showed that these sequences were more than 98.9% similar to each other but only distantly related to the previously recognized anaerobic ammonium oxidizer (below 91% similarity) and all other organisms represented in public 16S rRNA databases (similarities of below 79%). The retrieved sequences and the previously recognized anaerobic ammonium oxidizer represent two well-separated groups of a deep-branching lineage within the Planctomycetales. Quantitative FISH analysis with a newly designed specific probe showed that the novel bacterium, provisionally classified as "Candidatus Kuenenia stuttgartiensis" constituted the dominant fraction of the biofilm bacteria. In situ probing revealed that ammonia-oxidizing bacteria of the beta-subclass of Proteobacteria were also present, albeit in significant smaller amounts, within the anoxic biofilm. Comparative sequence analysis of a stretch of the gene encoding ammonia-monooxygenase (amoA) demonstrated the occurrence of the DNA of at least three different populations of beta-subclass ammonia oxidizers within the biofilm.
Neochlamydia hartmannellae gen. nov. sp. nov. (Parachlamydiaceae), an endoparasite of the amoeba Hartmannella vermiformis
2000 - Microbiology, 146: 1231-1239
Abstract:
Free-living amoebae are increasingly being recognized to serve as vehicles of dispersal for various bacterial human pathogens and as hosts for a variety of obligate bacterial endocytobionts. Several Chlamydia-like Acanthamoeba endocytobionts constituting the recently proposed family Parachlamydiaceae are of special interest as potential human pathogens. In this study coccoid bacterial endocytobionts of a Hartmannella vermiformis isolate were analysed. Infection of H. vermiformis with these bacteria resulted in prevention of cyst formation and subsequent host-cell lysis. Transfection experiments demonstrated that the parasites were not capable of propagating within other closely related free-living amoebae but were able to infect the distantly related species Dictyostelium discoideum. Electron microscopy of the parasites revealed typical morphological characteristics of the Chlamydiales, including the existence of a Chlamydia-like life-cycle, but indicated that these endocytobionts, in contrast to Chlamydia species, do not reside within a vacuole. Comparative 16S rRNA sequence analysis showed that the endocytobiont of H. vermiformis, classified as Neochlamydia hartmannellae gen. nov., sp. nov., is affiliated to the family Parachlamydiaceae. Confocal laser scanning microscopy in combination with fluorescence in situ hybridization using rRNA-targeted oligonucleotide probes confirmed the intracellular localization of the parasites and demonstrated the absence of other bacterial species within the Hartmannella host. These findings extend our knowledge of the phylogenetic diversity of the Parachlamydiaceae and demonstrate for the first time that these endocytobionts can naturally develop within amoebae of the genus Hartmannella.
Phylogenetic diversity among geographically dispersed Chlamydiales endosymbionts recovered from clinical and environmental isolates of Acanthamoeba spp
2000 - Appl. Environ. Microbiol., 66: 2613-2619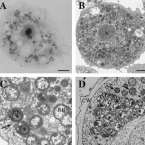
Abstract:
The recently proposed reorganization of the order Chlamydiales and description of new taxa are broadening our perception of this once narrowly defined taxon. We have recovered four strains of gram-negative cocci endosymbiotic in Acanthamoeba spp., representing 5% of the Acanthamoeba sp. isolates examined, which displayed developmental life cycles typical of members of the Chlamydiales. One of these endosymbiont strains was found stably infecting an amoebic isolate recovered from a case of amoebic keratitis in North America, with three others found in acanthamoebae recovered from environmental sources in North America (two isolates) and Europe (one isolate). Analyses of nearly full-length 16S rRNA gene sequences of these isolates by neighbor joining, parsimony, and distance matrix methods revealed their clustering with other members of the Chlamydiales but in a lineage separate from those of the genera Chlamydia, Chlamydophila, Simkania, and Waddlia (sequence similarities, <88%) and including the recently described species Parachlamydia acanthamoebae (sequence similarities, 91.2 to 93.1%). With sequence similarities to each other of 91.4 to 99.4%, these four isolates of intra-amoebal endosymbionts may represent three distinct species and, perhaps, new genera within the recently proposed family Parachlamydiaceae. Fluorescently labeled oligonucleotide probes targeted to 16S rRNA signature regions were able to readily differentiate two groups of intra-amoebal endosymbionts which corresponded to two phylogenetic lineages. These results reveal significant phylogenetic diversity occurring among the Chlamydiales in nontraditional host species and supports the existence of a large environmental reservoir of related species. Considering that all described species of Chlamydiales are known to be pathogenic, further investigation of intra-amoebal parachlamydiae as disease-producing agents is warranted.
Confocal scanning laser microscopy as a tool for the determination of 3D floc structure
1999 - Journal of Colloid and Interface Science, 220: 465-467Nitrogen loss in a nitrifying biofilm system
1999 - Wat. Sci. Tech., 39: 13-21
Use of microautoradiography and fluorescent in situ hybridization for characterization of microbial activity in activated sludge
1999 - Wat. Sci. Tech., 39: 1-9In situ detection of novel bacterial endosymbionts of Acanthamoeba spp. phylogenetically related to members of the Rickettsiales
1999 - Appl. Environ. Microbiol., 65: 206-212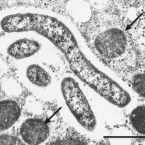
Abstract:
Acanthamoebae are ubiquitous soil and water bactivores which may serve as amplification vehicles for a variety of pathogenic facultative bacteria and as hosts to other, presently uncultured bacterial endosymbionts. The spectrum of uncultured endosymbionts includes gram-negative rods and gram-variable cocci, the latter recently shown to be members of the Chlamydiales. We report here the isolation from corneal scrapings of two Acanthamoeba strains that harbor gram-negative rod endosymbionts that could not be cultured by standard techniques. These bacteria were phylogenetically characterized following amplification and sequencing of the near-full-length 16S rRNA gene. We used two fluorescently labelled oligonucleotide probes targeting signature regions within the retrieved sequences to detect these organisms in situ. Phylogenetic analyses demonstrated that they displayed 99.6% sequence similarity and formed an independent and well-separated lineage within the Rickettsiales branch of the alpha subdivision of the Proteobacteria. Nearest relatives included members of the genus Rickettsia, with sequence similarities of approximately 85 to 86%, suggesting that these symbionts are representatives of a new genus and, perhaps, family. Distance matrix, parsimony, and maximum-likelihood tree-generating methods all consistently supported deep branching of the 16S rDNA sequences within the Rickettsiales. The oligonucleotide probes displayed at least three mismatches to all other available 16S rDNA sequences, and they both readily permitted the unambiguous detection of rod-shaped bacteria within intact acanthamoebae by confocal laser-scanning microscopy. Considering the long-standing relationship of most Rickettsiales with arthropods, the finding of a related lineage of endosymbionts in protozoan hosts was unexpected and may have implications for the preadaptation and/or recruitment of rickettsia-like bacteria to metazoan hosts.
Morphological and biochemical properties of Sphaerotilus sp. isolated from paper mill slimes
1999 - Appl. Environ. Microbiol., 65: 156-162Abstract:
Four strains of filamentous bacteria were isolated from slimes collected in different paper mill factories. Morphological and physiological characterization of the isolates indicated an affiliation with the genus Sphaerotilus. However, while the physiological properties of the isolates were almost identical, pronounced physiological differences between the isolates and Sphaerotilus natans DSM 6575(T), DSM 565, and DSM 566 with respect to their ability to metabolize complex polysaccharides, sugars, polyalcohols, or organic acids as carbon sources were detected. In contrast to the analyzed culture collection strains of S. natans, all paper mill isolates were able to grow at elevated temperatures of up to 40 degrees C. Comparative sequence analysis of nearly complete 16S ribosomal DNA (rDNA) sequences from the four new isolates demonstrated that the retrieved sequences were highly similar to each other (99.6 to 99.8% similarity) and to previously published partial 16S rDNA sequences of S. natans DSM 6575(T) and ATCC 15291. Polyphasic characterization of the isolated Sphaerotilus strains revealed interesting adaptations of the strains to the environmental paper mill conditions with regard to temperature tolerance and utilization of cellulose and starch.
Novel Bacterial Endosymbionts of Acanthamoeba isolates related to the Paramecium Symbiont Caedibacter caryophilus
1999 - Environ. Microbiol., 1: 357-367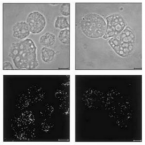
Abstract:
Acanthamoebae are increasingly being recognized as hosts for obligate bacterial endosymbionts, most of which are presently uncharacterized. In this study, the phylogeny of three Gram-negative, rod-shaped endosymbionts and their Acanthamoeba host cells was analysed by the rRNA approach. Comparative analyses of 16S rDNA sequences retrieved from amoebic cell lysates revealed that the endosymbionts of Acanthamoeba polyphaga HN-3, Acanthamoeba sp. UWC9 and Acanthamoeba sp. UWE39 are related to the Paramecium caudatum endosymbionts Caedibacter caryophilus, Holospora elegans and Holospora obtusa. With overall 16S rRNA sequence similarities to their closest relative, C. caryophilus, of between 87% and 93%, these endosymbionts represent three distinct new species. In situ hybridization with fluorescently labelled endosymbiont-specific 16S rRNA-targeted probes demonstrated that the retrieved 16S rDNA sequences originated from the endosymbionts and confirmed their intracellular localization. We propose to classify provisionally the endosymbiont of Acanthamoeba polyphaga HN-3 as 'Candidatus Caedibacter acanthamoebae', the endosymbiont of Acanthamoeba sp. strain UWC9 as 'Candidatus Paracaedibacter acanthamoebae' and the endosymbiont of Acanthamoeba sp. strain UWE39 as 'Candidatus Paracaedibacter symbiosus'. The phylogeny of the Acanthamoeba host cells was analysed by comparative sequence analyses of their 18S rRNA. Although Acanthamoeba polyphaga HN-3 clearly groups together with most of the known Acanthamoeba isolates (18S rRNA sequence type 4), Acanthamoeba sp. UWC9 and UWE39 exhibit <92% 18S rRNA sequence similarity to each other and to other Acanthamoeba isolates. Therefore, we propose two new sequence types (T13 and T14) within the genus Acanthamoeba containing, respectively, Acanthamoeba sp. UWC9 and Acanthamoeba sp. UWE39.
Phylogeny and in situ identification of a morphologically conspicuous bacterium Candidatus "Magnospira bakii" present at very low frequency in activated sludge
1999 - Environ. Microbiol., 1: 125-135Abstract:
A morphologically conspicuous bacterium that constituted a very small fraction (< 0.01%) of the total microbial community of activated sludge was enriched and analysed phylogenetically by a combination of cultivation-independent molecular and physical methods. The large, corkscrew-shaped, filamentous bacteria were first detected in municipal activated sludge by light microscopy owing to their unusual rotating gliding motility. Various attempts at microbiological enrichment and pure culture isolation with traditional techniques failed, as did attempts to retrieve the morphotype of interest by micromanipulation. In situ hybridization with the group-specific, rRNA-targeted oligonucleotide probe CF319a indicated a phylogenetic affiliation to the Cytophaga-Flexibacter group of the Cytophaga-Flavobacterium-Bacteroides phylum. Based on strong morphological resemblance to members of the genus Saprospira, additional 16S rRNA-targeted oligonucleotides with more narrow specificity were designed and evaluated for in situ hybridization to the morphotype of interest. Flow cytometric cell sorting based on the fluorescence conferred by probe SGR1425 and forward scatter enabled a physical enrichment of the helical coiled cells. Subsequent polymerase chain reaction (PCR) amplification of 16S rDNA fragments from whole fixed sorted cells with a primer pair based on probes CF319a and SGR1425 resulted in the retrieval of 12 almost identical partial 16S rDNA fragments with sequence similarities among each other of more than 99.2%. In situ hybridizations proved that the sequences that showed the highest similarity (88.4%) to the 16S rRNA of Saprospira grandis were indeed retrieved from the corkscrew-shaped filaments. The bacterium is likely to be a member of a genus of which no species has been cultured hitherto. It was consequently tentatively named 'Magnospira bakii' and has the taxonomic rank of Candidatus Magnospira bakii, as the ultimate taxonomic placement has to await its cultivation. In this study, it was demonstrated that even bacteria occurring at very low frequencies in highly complex environmental samples can be retrieved selectively without cultivation for further molecular analysis.
Probe EUB338 is insufficient for the detection of all Bacteria: Development and evaluation of a more comprehensive probe set
1999 - Syst. Appl. Microbiol., 22: 434-444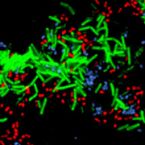
Abstract:
In situ hybridization with rRNA-targeted oligonucleotide probes has become a widely applied tool for direct analysis of microbial population structures of complex natural and engineered systems. In such studies probe EUB338 (AMANN et al., 1990) is routinely used to quantify members of the domain Bacteria with a sufficiently high cellular ribosome content. Recent reevaluations of probe EUB338 coverage based on all publicly available 16S rRNA sequences, however, indicated that important bacterial phyla, most notably the Planctomycetales and Verrucomicrobia, are missed by this probe. We therefore designed and evaluated two supplementary versions (EUB338-II and EUB338-III) of probe EUB338 for in situ detection of most of those phyla not detected with probe EUB338. In situ dissociation curves with target and non-target organisms were recorded under increasing stringency to optimize hybridization conditions. For that purpose a digital image software routine was developed. In situ hybridization of a complex biofilm community with the three EUB338 probes demonstrated the presence of significant numbers of probe EUB338-II and EUB338-III target organisms. The application of EUB338, EUB338-II and EUB338-III should allow a more accurate quantification of members of the domain Bacteria in future molecular ecological studies.
Diversity of sulfate-reducing bacteria in oxic and anoxic regions of a microbial mat characterized by comparative analysis of dissimilatory sulfite reductase genes
1999 - Appl. Environ. Microbiol., 65: 4666-4671Abstract:
Sequence analysis of genes encoding dissimilatory sulfite reductase (DSR) was used to identify sulfate-reducing bacteria in a hypersaline microbial mat and to evaluate their distribution in relation to levels of oxygen. The most highly diverse DSR sequences, most related to those of the Desulfonema-like organisms within the delta-proteobacteria, were recovered from oxic regions of the mat. This observation extends those of previous studies by us and others associating Desulfonema-like organisms with oxic habitats.
On the occurrence of anoxic microniches, denitrification, and sulfate reduction in aerated activated sludge
1999 - Appl. Environ. Microbiol., 65: 4189-4196Abstract:
A combination of different methods was applied to investigate the occurrence of anaerobic processes in aerated activated sludge. Microsensor measurements (O(2), NO(2)(-), NO(3)(-), and H(2)S) were performed on single sludge flocs to detect anoxic niches, nitrate reduction, or sulfate reduction on a microscale. Incubations of activated sludge with (15)NO(3)(-) and (35)SO(4)(2-) were used to determine denitrification and sulfate reduction rates on a batch scale. In four of six investigated sludges, no anoxic zones developed during aeration, and consequently denitrification rates were very low. However, in two sludges anoxia in flocs coincided with significant denitrification rates. Sulfate reduction could not be detected in any sludge in either the microsensor or the batch investigation, not even under short-term anoxic conditions. In contrast, the presence of sulfate-reducing bacteria was shown by fluorescence in situ hybridization with 16S rRNA-targeted oligonucleotide probes and by PCR-based detection of genes coding for the dissimilatory sulfite reductase. A possible explanation for the absence of anoxia even in most of the larger flocs might be that oxygen transport is not only diffusional but enhanced by advection, i.e., facilitated by flow through pores and channels. This possibility is suggested by the irregularity of some oxygen profiles and by confocal laser scanning microscopy of the three-dimensional floc structures, which showed that flocs from the two sludges in which anoxic zones were found were apparently denser than flocs from the other sludges.
Identification of some of the major groups of bacteria in efficient and nonefficient biological phosphorus removal activated sludge systems
1999 - Appl. Environ. Microbiol., 65: 4077-4084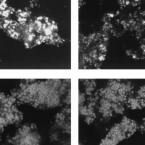
Abstract:
To investigate the bacteria that are important to phosphorus (P) removal in activated sludge, microbial populations were analyzed during the operation of a laboratory-scale reactor with various P removal performances. The bacterial population structure, analyzed by fluorescence in situ hybridization (FISH) with oligonucleotides probes complementary to regions of the 16S and 23S rRNAs, was associated with the P removal performance of the reactor. At one stage of the reactor operation, chemical characterization revealed that extremely poor P removal was occurring. However, like in typical P-removing sludges, complete anaerobic uptake of the carbon substrate occurred. Bacteria inhibiting P removal overwhelmed the reactor, and according to FISH, bacteria of the beta subclass of the class Proteobacteria other than beta-1 or beta-2 were dominant in the sludge (58% of the population). Changes made to the operation of the reactor led to the development of a biomass population with an extremely good P removal capacity. The biochemical transformations observed in this sludge were characteristic of typical P-removing activated sludge. The microbial population analysis of the P-removing sludge indicated that bacteria of the beta-2 subclass of the class Proteobacteria and actinobacteria were dominant (55 and 35%, respectively), therefore implicating bacteria from these groups in high-performance P removal. The changes in operation that led to the improved performance of the reactor included allowing the pH to rise during the anaerobic period, which promoted anaerobic phosphate release and possibly caused selection against non-phosphate-removing bacteria.
Combination of fluorescent in situ hybridization and microautoradiography - a new tool for structure-function analyses in microbial ecology
1999 - Appl. Environ. Microbiol., 65: 1289-1297
Nitrosospira sp. and Nitrospira sp. as dominant populations in a nitrifying fluidized bed reactor: Identification and activity in situ
1998 - Appl. Environ. Microbiol., 64: 3480-3485Combining fluorescent in situ hybridization (FISH) with cultivation and mathematical modeling to study population structure and function of ammonia-oxidizing bacteria in activated sludge
1998 - Wat. Sci. Tech., 37: 441-449In situ detection and identification of bacteria prior to their cultivation
1998 - Bioscience Microflora, 17: 15-22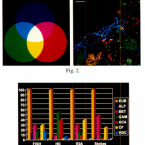
Semi-automated confocal laser scanning microscopy and image processing for the analysis of biofilms
1998 - Appl. Environ. Microbiol., 64: 4115-4127
Variability of Type 021N in activated sludge as determined by in situ substrate uptake pattern and in situ hybridization with fluorescent rRNA-targeted probes
1998 - Wat. Sci. Tech., 37: 423-430Monitoring the community structures of wastewater treatment plants: a comparison of old and new techniques
1998 - FEMS Microbiol. Ecol., 25: 205-216
Fluorescent in situ hybridization shows spatial distribution of yet uncultured Treponemes in biopsies from digital dermatitis lesions
1998 - Microbiology, 144: 2459-2467Abstract:
Fluorescence in situ hybridization (FISH) was performed on sections of plastic-embedded tissue using 16S rRNA-directed oligonucleotide probes to visualize uncultured treponemes in skin biopsies of cows with digital dermatitis. Plastic as embedding material allowed sectioning of hard and soft tissue with a defined thickness, avoiding the risk of dragging bacteria into the tissue while sectioning. furthermore, it provided a good signal-to-noise ratio. Using this method the spatial distribution of three different bacterial phylotypes was visualized simultaneously within the tissue. Whereas debris covering the ulcers contained a mixture of different micro-organisms, a layering of certain treponemal phylotypes was observed deeper in the epidermis. Confocal laser scanning microscopy and subsequent three-dimensional reconstruction of series of optical sections confirmed that the treponemes migrated intercellularly around the cells, most of them directed towards the dermis. In situ hybridization on tissue embedded in plastic proved to be a useful method to study mixed bacterial infections since it combines excellent histological conservation of tissue with identification of bacterial species by simultaneous use of probes labelled with different fluorescent dyes. This technique may have implications for in situ detection, identification and localization of microorganisms in veterinary as well as in human medicine.
Combined molecular and conventional analyses of nitrifying bacterium diversity in activated sludge: Nitrosococcus mobilis and Nitrospira-like bacteria as dominant populations
1998 - Appl. Environ. Microbiol., 64: 3042-3051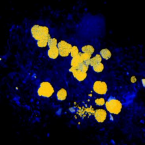
Abstract:
The ammonia-oxidizing and nitrite-oxidizing bacterial populations occurring in the nitrifying activated sludge of an industrial wastewater treatment plant receiving sewage with high ammonia concentrations were studied by use of a polyphasic approach. In situ hybridization with a set of hierarchical 16S rRNA-targeted probes for ammonia-oxidizing bacteria revealed the dominance of Nitrosococcus mobilis-like bacteria. The phylogenetic affiliation suggested by fluorescent in situ hybridization (FISH) was confirmed by isolation of N. mobilis as the numerically dominant ammonia oxidizer and subsequent comparative 16S rRNA gene (rDNA) sequence and DNA-DNA hybridization analyses. For molecular fine-scale analysis of the ammonia-oxidizing population, a partial stretch of the gene encoding the active-site polypeptide of ammonia monooxygenase (amoA) was amplified from total DNA extracted from ammonia oxidizer isolates and from activated sludge. However, comparative sequence analysis of 13 amoA clone sequences from activated sludge demonstrated that these sequences were highly similar to each other and to the corresponding amoA gene fragments of Nitrosomonas europaea Nm50 and the N. mobilis isolate. The unexpected high sequence similarity between the amoA gene fragments of the N. mobilis isolate and N. europaea indicates a possible lateral gene transfer event. Although a Nitrobacter strain was isolated, members of the nitrite-oxidizing genus Nitrobacter were not detectable in the activated sludge by in situ hybridization. Therefore, we used the rRNA approach to investigate the abundance of other well-known nitrite-oxidizing bacterial genera. Three different methods were used for DNA extraction from the activated sludge. For each DNA preparation, almost full-length genes encoding small-subunit rRNA were separately amplified and used to generate three 16S rDNA libraries. By comparative sequence analysis, 2 of 60 randomly selected clones could be assigned to the nitrite-oxidizing bacteria of the genus Nitrospira. Based on these clone sequences, a specific 16S rRNA-targeted probe was developed. FISH of the activated sludge with this probe demonstrated that Nitrospira-like bacteria were present in significant numbers (9% of the total bacterial counts) and frequently occurred in coaggregated microcolonies with N. mobilis.
Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration
1998 - J. Bacteriol., 180: 2975-2982
Abstract:
Microorganisms that use sulfate as a terminal electron acceptor for anaerobic respiration play a central role in the global sulfur cycle. Here, we report the results of comparative sequence analysis of dissimilatory sulfite reductase (DSR) genes from closely and distantly related sulfate-reducing organisms to infer the evolutionary history of DSR. A 1.9-kb DNA region encoding most of the alpha and beta subunits of DSR could be recovered only from organisms capable of dissimilatory sulfate reduction with a PCR primer set targeting highly conserved regions in these genes. All DNA sequences obtained were highly similar to one another (49 to 89% identity), and their inferred evolutionary relationships were nearly identical to those inferred on the basis of 16S rRNA. We conclude that the high similarity of bacterial and archaeal DSRs reflects their common origin from a conserved DSR. This ancestral DSR was either present before the split between the domains Bacteria, Archaea, and Eucarya or laterally transferred between Bacteria and Archaea soon after domain divergence. Thus, if the physiological role of the DSR was constant over time, then early ancestors of Bacteria and Archaea already possessed a key enzyme of sulfate and sulfite respiration.
In situ detection of a virulence factor mRNA and 16S rRNA in Listeria monocytogenes
1998 - FEMS Microbiol. Lett., 160: 159-168
Abstract:
Simultaneous in situ analysis of the structure and function of bacterial cells present within complex communities is a key for improving our understanding of microbial ecology. A protocol for the in situ identification of Listeria spp. using fluorescently tagged, rRNA-targeted oligonucleotide probes was developed. Ethanol fixation and enzymatic pretreatment with lysozyme and proteinase K were used to optimize whole cell hybridization of exponential phase and stationary phase Listeria spp. cells. In parallel, transcript probes carrying multiple digoxigenin molecules were combined with anti-digoxigenin Fab antibody fragments labeled with horseradish peroxidase to detect, via the catalytic deposition of fluorescein-tyramide, the iap-mRNA in single Listeria monocytogenes cells. The iap gene encodes the associated virulence factor p60. Application of the new signal amplification technique resulted in strong signals comparable in intensity to those obtained with fluorescently labeled rRNA-targeted oligonucleotide probes.
In situ identification of nocardioform actinomycetes in activated sludge using fluorescent rRNA-targeted oligonucleotide probes
1998 - Microbiology, 144: 249-259Abstract:
Hitherto, few environmental samples have been investigated by a 'full cycle rRNA analysis'. Here the results of in situ hybridization experiments with specific rRNA-targeted oligonucleotide probes developed on the basis of new sequences derived from a previously described comparative 16S rRNA analysis of nocardioform actinomycetes in activated sludge are reported. Application of the specific probes enabled identification and discrimination of the distinct populations of nocardioform actinomycetes in activated sludge. One of the specific probes (DLP) detected rod-shaped bacteria which were found in 13 of the 16 investigated sludge samples from various wastewater treatment plants, suggesting their importance in the wastewater treatment process. Another probe (GLP2) hybridized with typically branched filaments of nocardioforms mainly found in samples from enhanced biological phosphorus removal plants, suggesting that these bacteria are involved in sludge foaming. The combination of in situ hybridization with fluorescently labelled rRNA-targeted oligonucleotide probes and confocal laser scanning microscopy improved the detection of nocardioform actinomycetes, which often showed only weak signals inside the activated-sludge flocs.
Phylogenetic analysis of microbial communities associated with two deep, anaerobic, alkaline aquifers
1997 - Appl. Environ. Microbiol., 63: 1498-1504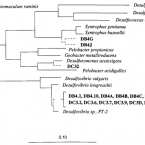
In situ analysis of nitrifying bacteria in sewage treatment plants
1996 - Wat. Sci. Tech., 34: 237-244
Phylogenetic probes for analyzing abundance and spatial organization of nitrifying bacteria
1996 - Appl. Environ. Microbiol., 62: 2156-2162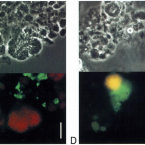
Abstract:
A hierarchical set of five 16S rRNA-targeted oligonucleotide DNA probes for phylogenetically defined groups of autotrophic ammonia- and nitrite-oxidizing bacteria was developed for environmental and determinative studies. Hybridization conditions were established for each probe by using temperature dissociation profiles of target and closely related nontarget organisms to document specificity. Environmental application was demonstrated by quantitative slot blot hybridization and whole-cell hybridization of nitrifying activated sludge and biofilm samples. Results obtained with both techniques suggested the occurrence of novel populations of ammonia oxidizers. In situ hybridization experiments revealed that Nitrobacter and Nitrosomonas species occurred in clusters and frequently were in contact with each other within sludge flocs.
Characterization of bacterial communities from activated sludge: Culture-dependent numerical identification versus in situ identification using group- and genus-specific rRNA-targeted oligonucleotide probes
1996 - Microb. Ecol., 32: 101-121Abstract:
The structures of bacterial communities were studied in activated sludge samples obtained from the aerobic and anaerobic zones of a wastewater treatment plant showing enhanced phosphorous removal. Samples were analyzed by in situ hybridization with oligonucleotide probes complementary to selected regions of the 16S and 23S ribosomal RNA (rRNA) characteristic for defined phylogenetic entities (genera and larger groups). The microbial community structures revealed by molecular techniques were compared with the compositions of culturable bacterial communities, obtained from the characterization of 255 isolates from tryptone-soy (TS) agar and R2A agar. These isolates were characterized by 89 physiological tests and their cellular fatty acid patterns, and identified. Culture-dependent techniques indicated the following distribution: different Aeromonas spp. (2.7-8.3% on R2A agar; 45.0-63.7% on TS agar), Acinetobacter spp. (5.4-9.0% on R2A agar; 5.0-9.1% on TS agar), Pseudomonas spp. (up to 10% on R2A agar) and Shewanella putrefaciens (up to 3.0% on R2A agar), all members of the gamma subclass of Proteobacteria, were isolated most frequently. The relatively rare isolates of the beta subclass were identified as Acidovorax spp., Alcaligenes spp., and Comamonas spp.. The Gram-positive bacteria (high DNA G+C) were assigned mainly to Arthrobacter spp., Microbacterium spp., and Mycobacterium phlei. In order to assess the in situ abundance of the most frequently isolated genus, Aeromonas, two rRNA-targeted oligonucleotide probes were developed. The two gamma proteobacterial genera Aeromonas and Acinetobacter constituted less than 5% of all bacteria. In situ, Proteobacteria belonging to the beta subclass and high G+C Gram-positive bacteria were dominant. From filamentous bacteria, Sphaerotilus spp. and Leptothrix spp. could be detected occasionally. In addition, one sample contained a high proportion of the morphologically distinct filaments of Microthrix parvicella.
In situ visualization of high genetic diversity in a natural microbial community
1996 - J. Bacteriol., 178: 3496-3500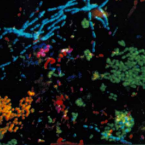
Abstract:
Simultaneous in situ visualization of seven distinct bacterial genotypes, all affiliated with the phylogenetically narrow group of beta-1 Proteobacteria, was achieved in activated sludge. This finding indicates that the high diversity found in the same sample by direct rRNA sequence retrieval was indeed present in this complex community. By the combination of comparative rRNA sequence analysis, in situ hybridization with fluorescently labeled, rRNA-targeted oligonucleotides and confocal laser scanning microscopy several microbial populations can be analyzed for abundance, relative spatial distribution and phylogeny directly at their site of action without prior cultivation.
In situ identification of ammonia-oxidizing bacteria
1995 - Syst. Appl. Microbiol., 17: 251-264
The abundance of Zoogloea ramigera in sewage treatment plants
1995 - Appl. Environ. Microbiol., 61: 702-707
Abstract:
Zoogloea ramigera has long been considered the typical activated sludge bacterium responsible for the formation of activated sludge flocs. On the basis of the results of a comparative sequence analysis, we designed three oligonucleotide probes complementary to characteristic regions of the 16S rRNAs of Z. ramigera ATCC 19544T (T = type strain) and two misclassified strains, Z. ramigera ATCC 25935 and ATCC 19623. Dissociation temperatures were determined, and probe specificities, as well as the potential of probes for whole-cell hybridization, were evaluated by using numerous reference organisms. Several activated sludge samples were examined with these probes by using both the in situ and dot blot hybridization methods. Only the type strain probe hybridized to cells that accumulated in the typical branched gelatinous matrices, the so-called Zoogloea fingers. This probe revealed cells in most of the activated sludge samples studied. We found that relatively high levels of Z. ramigera cells (up to approximately 10% of the total number of cells) and typical morphology tended to be linked to overloading of sewage plants. The probe directed to rejected type strain Z. ramigera ATCC 19623 bound to only a few cells. Cells that reacted with the probe complementary to Z. ramigera ATCC 25935, which was originally isolated from a trickling filter, were not observed in activated sludge.
Identification and in situ detection of gram-negative filamentous bacteria in activated sludge
1994 - Syst. Appl. Microbiol., 17: 405-417In situ characterization of the microbial consortia active in two wastewater treatment plants
1994 - Wat. Res., 28: 1715-1723
Probing activated sludge with fluorescently labeled rRNA targeted oligonucleotides
1994 - Wat. Sci. Tech., 29: 15-23In situ probing of Gram-positive bacteria with high DNA G+C content using 23S rRNA-targeted oligonucleotides
1994 - Microbiology, 140: 2849-2858Abstract:
23S-rRNA-targeted oligonucleotide probes were designed for the phylogenetic group 'Gram-positive bacteria with high G + C content of DNA' (GPBHGC). A sequence idiosyncrasy in two adjacent base pairs in the stem of helix 69 in domain IV of the 23S rRNA is present in all hitherto analysed strains of GPBHGC. An oligonucleotide probe targeted to this region hybridized only with strains of GPBHGC and was successfully used for in situ monitoring of these cells in activated sludge. Another unique feature of the 23S rRNA molecules of GPBHGC is a large insertion in domain III. Fluorescent oligonucleotides targeted to the highly variable regions of the rRNA within the insertions of Corynebacterium glutamicum DSM 20300, Aureobacterium testaceum DSM 20166 and Brevibacterium sp. DSM 20165 hybridized specifically to their target strains, whereas probing with oligonucleotides complementary to the rRNA-coding strand of the 23S rDNA and to the spacer between 16S and 23S rRNA of C. glutamicum did not result in detectable fluorescence. This confirmed that the large 23S insertions are indeed present in 23S rRNAs of GPBHGC and provide potential target sites for highly specific nucleic acid probes.
In situ analysis of microbial consortia in activated sludge using fluorescently labelled, rRNA-targeted oligonucleotide probes and confocal scanning laser microscopy
1994 - J. Microscopy, 176: 181-187Abstract:
Activated sludge flocs are complex consortia of various micro-organisms. The community structures of samples taken from municipal sewage treatment plants were characterized using fluorescently labelled, 16S and 23S rRNA-targeted oligonucleotide probes in combination with confocal scanning laser microscopy (CSLM). In comparison with conventional epifluorescence microscopy, CSLM considerably improved the capability to visualize directly the spatial distribution of defined bacterial populations inside the sludge flocs. Analyses could be performed at high resolution undisturbed by problems such as autofluorescence or limited spatial resolution in thick samples. In addition, CSLM was used to analyse some structural properties of paraformaldehyde-fixed activated sludge flocs, such as floc size and homogeneity. Typical floc sizes were found to be in the range between 5 and 50 microns. Whereas most of the flocs were completely colonized by bacteria, there were also examples of flocs containing gas bubbles or particles in the interior.
Development of an rRNA-targeted oligonucleotide probe specific for the genus Acinetobacter and its application for in situ monitoring in activated sludge
1994 - Appl. Environ. Microbiol., 60: 792-800
Abstract:
Enhanced biological phosphate removal in an anaerobic-aerobic activated sludge system has generally been ascribed to members of the genus Acinetobacter. A genus-specific 16S rRNA-targeted oligonucleotide probe was developed to investigate the role ofAcinetobacter spp. in situ. Nonisotopic dot blot hybridization to 66 reference strains, including the seven described Acinetobacter spp., demonstrated the expected probe specificity. Fluorescent derivatives were used for in situ monitoring of Acinetobacter spp. in the anaerobic and aerobic compartments of a sewage treatment plant with enhanced biological phosphate removal. Microbial community structures were further analyzed with oligonucleotide probes specific for the alpha, beta, or gamma subclasses of the class Proteobacteria, for the Cytophaga-Flavobacterium cluster, for gram-positive bacteria with a high G+C DNA content, and for all bacteria. Total cell counts were determined by 4',6-diamidino-2-phenylindole staining. In both the anaerobic and the aerobic basins, the activated sludge samples were dominated by members of the class Proteobacteria belonging to the beta subclass and by gram-positive bacteria with a high G+C DNA content. Acinetobacter spp. constituted less than 10% of all bacteria. For both basins, the microbial community structures determined with molecular techniques were compared with the compositions of the heterotrophic saprophytic microbiota determined with agar plating techniques. Isolates on nutrient-rich medium were classified by whole-cell hybridization with rRNA-targeted probes and fatty acid analysis. Cultivation on nutrient-rich medium favored the growth of members of the gamma subclass of Proteobacteria and selected against the growth of members of the beta subclass of Proteobacteria and gram-positive bacteria with a high G+C DNA content; 33% of the cultured bacteria from the anaerobic basin and 32% from the aeration basin were identified as Acinetobacter spp. The addition of small amounts of iron salts for chemical phosphate precipitation had no influence on the constitution of the microbial consortia. Enrichment of the return sludge with 20 mg of acetic acid per liter for 3 days significantly increased the relative abundance of gram-positive bacteria with a high G+C DNA content but had no effect on the numbers of Acinetobacter spp. The dominance of gram-positive bacteria with a high G+C DNA content and the presence of polyphosphate inclusions in these bacteria indicate that they may play a major role in biological phosphate removal.
Probing activated sludge with oligonucleotides specific for proteobacteria: inadequacy of culture-dependent methods for describing microbial community structure
1993 - Appl. Environ. Microbiol., 59: 1520-1525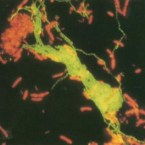
Abstract:
Bacterial community structures in activated sludge samples from aeration tanks of a two-stage system with a high-load first stage and a low-load second stage were analyzed with oligonucleotide probes. The probes were complementary to conserved regions of the rRNA of the alpha, beta, and gamma subclasses of proteobacteria and of all bacteria. Group-specific cell counts were determined by in situ hybridization with fluorescent probe derivatives. Contributions of the proteobacterial subclasses to total bacterial rRNA were quantified by dot blot hybridization with digoxigenin-labeled oligonucleotides. The activated sludge samples were dominated by proteobacteria from the alpha, beta, or gamma subclass. These proteobacteria account for about 80% of all active bacteria found in the activated sludge. For both samples the community structures determined with molecular techniques were compared with the composition of the heterotrophic saprophyte flora isolated on nutrient-rich medium. Probes were used to rapidly classify the isolates and to directly monitor population shifts in nutrient-amended, activated sludge samples. The rich medium favored growth of gamma-subclass proteobacteria (e.g., enterobacteria) and selected against beta-subclass proteobacteria. The culture-dependent community structure analysis of activated sludge produced partial and heavily biased results. A more realistic view will be obtained by using in situ techniques.
Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria: problems and solutions
1992 - Syst. Appl. Microbiol, 15: 593-600
Abstract:
Based on comparative analyses of 16S and 23S ribosomal RNA sequences we have located sites specific for the alpha-, beta-, and gamma-subclasses of Proteobacteria. Short oligodeoxynucleotides complementary to these signature regions were evaluated as potential nucleic acid probes for the differentiation of the major subclasses of Proteobacteria. Hybridization conditions were optimized by the addition of form-amide to the hybridization buffer and high stringency post-hybridization washing. Single-mismatch discrimination of probes was further improved by blocking nontarget probe binding sites with competitor oligonucleotides. Nonisotopic dot-blot hybridization to reference strains demonstrated the expected probe specificities, whole cell hybridization with fluorescent probe derivatives allowed the classification of individual microbial cells. The probes will be useful for determinative studies and for the in situ monitoring of population distribution and dynamics in microbial communities.
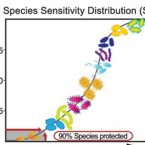

Book chapters and other publications
Mikrobiome – Wissensstand und Perspektiven
2019 - 17-29. in Die unbekannte Welt der Mikrobiome; Rundgespräche Forum Ökologie Bd. 47. (Bauer J & von Mutius E). Bayerische Akademie der Wissenschaften; Verlag Dr. Friedrich Pfeil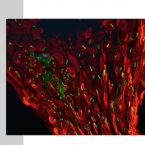
Nitrospira
2018 - Trends Microbiol., 5: 462-463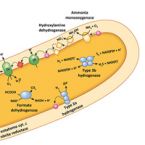
Abstract:
In this infographic, the key metabolic functions of Nitrospira and the role that these bacteria play in nitrification and other processes in the environment is shown. Nitrospira plays pivotal roles in nitrification as an aerobic chemolithoautotrophic nitrite-oxidizing bacterium. These bacteria often occur in close association with ammonia-oxidizing bacteria or archaea that convert ammonia to nitrite, which is further oxidized to nitrate by Nitrospira. However, in 'reciprocal feeding' interactions, Nitrospira can also provide ammonia oxidizers with ammonia released from urea or cyanate, which is further nitrified as described above. Recently discovered Nitrospira members even catalyze both nitrification steps alone and are therefore called complete ammonia oxidizers or 'comammox' organisms. Some strains of Nitrospira utilize alternative substrates, such as H and formate, using oxygen or nitrate as terminal electron acceptor, and can exploit these energy sources concurrently with aerobic nitrite oxidation. This metabolic versatility enables Nitrospira to colonize a broad range of habitats and to sustain shifts in environmental conditions such as changing oxygen concentrations.
Candidatus Nitrosotenuaceae
2016 - 1-5. in Bergey’s Manual of Systematics of Archaea and Bacteria. (William B. Whitman)). John Wiley & Sons, Chichester, England
Abstract:
Candidatus Nitrosotenuaceae is a family of Thaumarchaeota that consists of a single genus, Ca. Nitrosotenuis, which can be found widely distributed in soils, freshwater, hot springs, the subsurface, and activated sludge. They may be rods or spheres and may or may not have flagella. Similar to other known and described Thaumarchaeota, Ca. Nitrosotenuaceae are aerobic chemolithautotrophs that use energy gained from the oxidation of ammonia to nitrite to fix carbon via modified 3-hydroxypropionate/4 –hydroxybutyrate carbon fixation pathway. At this time, no pure culture of Ca. Nitrosotenuaceae exists, but some of the six available enrichments with members of the genus are almost pure. It remains to be demonstrated whether the remaining bacterial contaminants provide essential compounds to Ca. Nitrosotenuaceae. Enrichments of Ca. Nitrosotenuaceae are intolerant to high salinity (>0.3%), although they are phylogenetically related to the Group I.1a Thaumarchaeota (Ca. Nitrosopumilaceae), which includes taxa that are widely distributed in the ocean.
Candidatus Nitrosotenuis
2016 - 1-9. in Bergey’s Manual of Systematics of Archaea and Bacteria. (William B. Whitman). John Wiley & Sons, Chichester, England
Abstract:
Candidatus Nitrosotenuis is a genus of Thaumarchaeota that can be found widely distributed in soils, freshwater, hot springs, the subsurface, and activated sludge. They may be rods or spheres and may or may not have flagella. Like other known and described Thaumarchaeota, Ca. Nitrosotenuis are aerobic chemolithautotrophs that use energy gained from the oxidation of ammonia to nitrite to fix carbon via modified 3-hydroxypropionate/4-hydroxybutyrate carbon fixation pathway. At this time, no pure culture of Ca. Nitrosotenuis exists, but some of the six available enrichments with members of the genus are almost pure. It remains to be demonstrated whether the remaining bacterial contaminants provide essential compounds to Ca. Nitrosotenuis. Enrichments of Ca. Nitrosotenuis are intolerant to high salinity (>0.3%), although they are phylogenetically related to the Group I.1a Thaumarchaeota (Nitrosopumilaceae), which includes taxa that are widely distributed in the ocean.
-
The microbiology of nitrogen removal
2010 - 259-280. in The microbiology of activated sludge. (Nielsen PH, Seviour RJ). IWA Publishing, London, UK.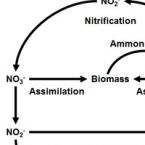
Diversity, environmental genomics, and ecophysiology of nitrite-oxidizing bacteria
2010 - 295-322. in Nitrification. (Klotz MG, Ward BB, Arp DJ). ASM Press, Washington, DC
Microarrays for studying the composition and function of microbial communities
2010 - 397-411. in The microbiology of activated sludge. (Nielsen PH, Seviour RJ). IWA Publishing, London, UK
Evolution and ecology of microbes dissimilating sulfur compounds: Insights from siroheme sulfite reductases
2008 - 46-59. in Microbial Sulfur Metabolism. (Friedrich C, Dahl C). Springer, New York
Abstract:
Sulfur microorganisms have been thriving on Earth since the dawn of life and are still of central importance for the functioning of modern ecosystems. Here, we summarize the current perception of the evolution of dissimilatory siroheme sulfite reductases (DSRs), antique key enzymes in the energy metabolism of sulfur microbes. We further give recent examples of the diversity and ecology of uncultured sulfur-dissimilating microorganisms; unprecedented insights that were only made possible by exploiting DSR-encoding genes as molecular markers in environmental surveys.
Molecular strategies for studies of natural populations of sulphate-reducing microorganisms
2007 - 39-117. in Sulfate-reducing bacteria: Environmental and engineered systems. (Barton LL, Hamilton WA). Cambridge University Press, Cambridge, UK
Journal Club - A microbial ecologist discovers another benefit of sex
2006 - Nature, 443: 485Crenothrix polyspora - ein ungewöhnlicher Methanoxidierer
2006 - BioSpektrum, 7: 718-720
Applications of nucleic acid microarrays in soil microbial ecology
2006 - 18-41. in Molecular approaches to soil, rhizosphere and plant microorganism analysis. (Cooper JE, Rao JR). CABI Publishing, Wallingford, Oxfordshire, UK
Abstract:
This chapter introduces the general methodological principles of DNA microarrays and highlight crucial steps in developing and applying this hybridization format for the analysis of complex microbial communities in the environment. Special emphasis is given to soil habitats. Microarrays for microbial community analysis have been classified into three main categories depending on the combination of probe types and target molecules exploited: (i) community genome arrays; (ii) rRNA-based oligonucleotide microarrays (PhyloChips, phylogenetic oligonucleotide arrays); and (iii) functional gene arrays. Benefits and caveats of these three microarrays are illustrated and discussed. The application of microarray technology is not restricted to identification of genes/microorganisms in the environment, but additionally has great potential for ecophysiological characterization of microbial populations.
Environmental chlamydia genomics
2006 - 25-44. in Chlamydia: genomics and pathogenesis. (Bavoil PM, Wyrick PB). Horizon Scientific Press, Norfolk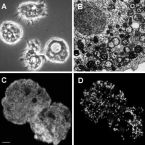
Abstract:
The discovery of chlamydia-related bacteria in the environment dramatically changed our perception of chlamydial diversity and their environmental occurrence. These so-called environmental chlamydiae display the unique chlamydial developmental cycle and mainly live as symbionts of free-living amoebae. Although there are some indications that environmental chlamydiae might also be able to infect humans, their pathogenic potential is still unclear. The first complete genome analysis of an environmental chlamydia strain, Protochlamydia amoebophila UWE25, provided novel insights into its biology, which, because of its larger genome compared to the Chlamydiaceae, opened a window on the genetic make-up of the chlamydial ancestor and the evolution of chlamydial virulence. The last common ancestor of the environmental chlamydiae and Chlamydiaceae was already adapted to intracellular survival in early eukaryotes, but was less dependent on its host cell. Several virulence factors required by Chlamydiaceae for interaction with their hosts evolved from genes of the last common ancestor of the environmental chlamydiae and the Chlamydiaceae.
The knowledge explosion in environmental microbiology offers new opportunities in biotechnology
2006 - Curr. Opin. Biotechnol., 17: 227-228Crystal ball: The community level: physiology and interactions of prokaryotes in the wilderness
2005 - Environ. Microbiol., 7: 472-485Fluorescence in situ hybridization for the detection of prokaryotes
2005 - 213-239. in Advanced Methods in Molecular Microbial Ecology. (Osborn AM, Smith CJ). Bios-Garland, Abingdon, UK
Isotopic-labelling methods for deciphering the function of uncultured micro-organisms
2005 - 1-10. in SGM symposium 65: micro-organisms and earth systems - advances in geomicrobiology. (Gadd GM, Semple KT, Lappin-Scott HM). Cambridge University Press
Microbial ecology of activated sludge
2005 - Microbiology Today, February 2005, 29-31
Die Evolution der Chlamydien - Einblicke in die Entwicklungsgeschichte bedeutender bakterieller Krankheitserreger
2005 - GenomXpress Sonderausgabe, 3: 50
Die Evolution der Chlamydien - Einblicke in die Entwicklungsgeschichte bedeutender bakterieller Krankheitserreger aus einer genomischen Perspektive
2004 - GenomXPress, 2: 13-14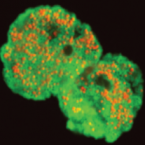
Analyse komplexer mikrobieller Lebensgemeinschaften mit phylogenetischen Mikroarrays
2004 - BioSpektrum, 6: 745-747
The lithoautotrophic ammonia oxidizers
2003 - 778-811. in The Prokaryotes: An evolving electronic resource for the microbiological community; 3rd edition, release 3.13, March 2003. Print version: 3rd edition, vol. 5, 2006. (Schleifer KH, Dworkin M, Falkow S, Rosenberg E, Stackebrandt E). Springer Verlag, New York
Halbautomatische und kultivierungs-unabhängige Quantifizierung von Bakterien in komplexen Umweltproben
2002 - Laborwelt, 1: 10-14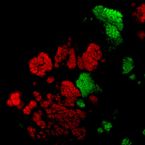
Activated Sludge - Molecular techniques for determining community composition
2002 - 26-43. in The Encyclopedia of Environmental Microbiology. (Bitton G). Wiley - New York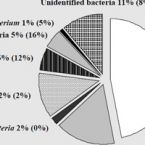
Molekulare Funktionsanalyse von Mikroorganismen
2002 - 41-54. in Rundgespräche der Kommission für Ökologie, Bd. 23 "Bedeutung der Mikroorganismen für die Umwelt". Verlag Dr. Friedrich Pfeil, MünchenFilamentous bacteria in activated sludge: Current taxonomic status and ecology
2002 - 1287-1306. in The Encyclopedia of Environmental Microbiology. (Bitton G). Wiley, New YorkDasein im Verborgenem. Bakterien, die in Acanthamöben leben
2001 - Biologie in unserer Zeit, 31: 160-168
Oxidation of inorganic nitrogen compounds as an energy source
2001 - 457-495. in The Prokaryotes vol. 2 (electronic version; printed version of this chapter appeared in 2006). (Dworkin M). Springer Verlag, New York
Abbau von Stickstoffverbindungen in Kläranlagen: Neue Nitrifikanten in Kläranlagen entdeckt
2000 - BioSpektrum, 3: 186-188
Die Anwendung von in situ-Hybridisierung zur Identifizierung der an der Stickstoff- und Phosphorentfernung beteiligten Bakterien in Kläranlagen
1998 - 52: 25-41. in Neueste Entwicklungen in der in situ- Charakterisierung mikrobieller Biozönosen in Abwasser, Oberflächengewässern, Grund- und Trinkwasser. Münchner Beiträge zur Abwasser-, Fischerei- und Flußbiologie.. Oldenbourg Verlag, München, Wien
In situ-Bestimmung fädiger Belebtschlammbakterien zur frühzeitigen Beurteilung von Blähschlamm und Schwimmschlammentwicklung
1998 - 52: 61-72. in Neueste Entwicklungen in der in situ- Charakterisierung mikrobieller Biozönosen in Abwasser, Oberflächengewässern, Grund- und Trinkwasser. Münchner Beiträge zur Abwasser-, Fischerei- und Flußbiologie.. Oldenbourg Verlag, München, Wien
3-D analysis of complex microbial communities by combining confocal laser scanning microscopy and fluorescence in situ hybridization (FISH)
1998 - 467-486. in Digital Image Analysis of Microbes.. (Wilkinson MHF, Schut F). John Wiley & Sons, Chichester, England
Molecular techniques for determining microbial community structures in activated sludge
1997 - 61-71. in Microbial community analysis: The key to the design of biological wastewater treatment systems.. (Cloete TE, Muyima NYO). University Press, Cambridge
In-situ-Nachweis der untergeordneten Rolle von Bakterien der Gattung Acinetobacter bei der erhöhten biologischen P-Elimination
1995 - GWF Wasser Abwasser, 136: 17-23
In situ identification of nonculturable bacteria with rRNA-targeted nucleic acid probes
1993 - 515-520. in Trends in Microbial Ecology. (Guerrero R, Pedros-Alio C). Spanish Society for Microbiology, Barcelona, SpainIn-situ-Nachweis der untergordneten Rolle von Acinetobacter in Kläranlagen mit biologisch erhöhter Phosphateliminierung
1993 - 91-100. in Wechselwirkungen der biologischen und chemischen Phosphorelimination.. (Hahn HH, Trauth R). Veröff. Inst. Siedlungswasserbau, Universität Karlsruhe
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at