Publications
Publications in peer reviewed journals
The symbiotic 'all-rounders': Partnerships between marine animals and chemosynthetic nitrogen-fixing bacteria.
2020 - Appl Environ Microbiol, in press
Abstract:
Nitrogen fixation is a widespread metabolic trait in certain types of microorganisms called diazotrophs. Bioavailable nitrogen is limited in various habitats on land and in the sea, and accordingly, a range of plant, animal, and single-celled eukaryotes have evolved symbioses with diverse diazotrophic bacteria, with enormous economic and ecological benefits. Until recently, all known nitrogen-fixing symbionts were heterotrophs such as nodulating rhizobia, or photoautotrophs such as cyanobacteria. In 2016, the first chemoautotrophic nitrogen-fixing symbionts were discovered in a common family of marine clams, the Lucinidae. Chemosynthetic nitrogen-fixing symbionts use the chemical energy stored in reduced sulfur compounds to power carbon and nitrogen fixation, making them metabolic 'all-rounders' with multiple functions in the symbiosis. This distinguishes them from heterotrophic symbionts that require a source of carbon from their host, and their chemosynthetic metabolism distinguishes them from photoautotrophic symbionts that produce oxygen, a potent inhibitor of nitrogenase. In this review, we consider evolutionary aspects of this discovery, by comparing strategies that have evolved for hosting intracellular nitrogen-fixing symbionts in plants and animals. The symbiosis between lucinid clams and chemosynthetic nitrogen-fixing bacteria also has important ecological impacts, as they form a nested symbiosis with endangered marine seagrasses. Notably, nitrogen fixation by lucinid symbionts may help support seagrass health by providing a source of nitrogen in seagrass habitats. These discoveries were enabled by new techniques for understanding the activity of microbial populations in natural environments. However, an animal (or plant) host represents a diverse landscape of microbial niches due to its structural, chemical, immune and behavioural properties. In future, methods that resolve microbial activity at the single cell level will provide radical new insights into the regulation of nitrogen fixation in chemosynthetic symbionts, shedding new light on the evolution of nitrogen-fixing symbioses in contrasting hosts and environments.
A novel alphaproteobacterium with a small genome identified from the digestive gland of multiple species of abalone.
2020 - Environ Microbiol Rep, 4: 387-395
Abstract:
We identified an alphaproteobacterium in the digestive gland of the abalone species Haliotis discus hannai. This phylotype dominated our 16S rRNA clone libraries from the digestive gland of H. discus hannai. Diversity surveys revealed that this phylotype was associated with H. discus hannai and also in another host species, H. gigantea. Whole genome phylogenies placed this bacterium as a new member affiliated with the family Rhodospirillaceae in Alphaproteobacteria. Gene annotation revealed a nearly complete glycolysis pathway but no TCA cycle, but the presence of anaerobic ribonucleoside-triphosphate reductase and oxygen-insensitive NAD(P)H-dependent nitroreductase, which show the genomic potential for anaerobic metabolism. A large cluster of genes encoding ankyrin repeat proteins (ANK) of eukaryotic-like repeat domains and a large gene set for the flagellar system were also detected. Alginate-binding periplasmic proteins and key genes responsible for alginate assimilation were found in the genome, which could potentially contribute to the breakdown of the host's alginate-rich macroalgal diet. These results raise the possibility that this novel alphaproteobacterium is a widespread member of the abalone microbiome that may use polysaccharides derived from its host's macroalgal diet.
Virulence characterization and comparative genomics of Listeria monocytogenes sequence type 155 strains.
2020 - BMC Genomics, 21: 847
Abstract:
Listeria (L.) monocytogenes strains show a high diversity regarding stress tolerance and virulence potential. Genome studies have mainly focused on specific sequence types (STs) predominantly associated with either food or human listeriosis. This study focused on the prevalent ST155, showing equal distribution among clinical and food isolates. We evaluated the virulence potential of 20 ST155 strains and performed comparative genomic analysis of 130 ST155 strains isolated from food, food processing environments and human listeriosis cases in different countries and years.
The in vitro virulence assays using human intestinal epithelial Caco2 and hepatocytic HEPG2 cells showed an impaired virulence phenotype for six of the 20 selected ST155 strains. Genome analysis revealed no distinct clustering of strains from the same source category (food, food processing environment, and clinical isolates). All strains harbored an intact inlA and inlB locus, except four strains, which had an internal deletion in the inlA gene. All strains harbored LIPI-1, but prfA was present in a longer variant in six strains, all showing impaired virulence. The longer PrfA variant resulted in lower expression of inlA, inlB, and prfA, and no expression of hly and actA. Regarding stress-related gene content, SSI-1 was present, whereas qacH was absent in all strains. 34.6% of the strains harbored a plasmid. All but one ST155 plasmids showed high conservation and harbored cadA2, bcrABC, and a triphenylmethane reductase.
This study contributes to an enhanced understanding of L. monocytogenes ST155 strains, being equally distributed among isolates from humans, food, and food processing environments. The conservation of the present genetic traits and the absence of unique inherent genetic features makes these types of STs especially interesting since they are apparently equally adapted to the conditions in food processing environments, as well as in food as to the human host environment. However, a ST155-specific mutation resulting in a longer PrfA variant impaired the virulence potential of several ST155 strains.Co-infection of chicken layers with Histomonas meleagridis and avian pathogenic Escherichia coli is associated with dysbiosis, cecal colonization and translocation of the bacteria from the gut lumen
2020 - Front Microbiol, 11: 586437
Abstract:
Histomonosis in chickens often appears together with colibacillosis in the field. Thus, we have experimentally investigated consequences of the co-infection of birds with Histomonas meleagridisand avian pathogenic Escherichia coli (APEC) on the pathology, host microbiota and bacterial translocation from the gut. Commercial chicken layers were infected via oral and cloacal routes with lux-tagged APEC with or without H. meleagridis whereas negative controls were left uninfected. Except one bird, which died due to colibacillosis, no clinical signs were recorded in birds infected with bioluminescence lux gene tagged E. coli. In co-infected birds, depression and ruffled feathers were observed in 4 birds and average body weight gain significantly decreased. Typhlitis caused by H. meleagridis was present only in co-infected birds, which also had pronounced microscopic lesions in systemic organs such as liver, heart and spleen. The 16S rRNA gene amplicon sequencing showed that in co-infected birds, corresponding to the severity of cecal lesions, microbial species richness and diversity in caeca greatly decreased and the abundance of the Escherichia group, Helicobacter and Bacteroides was relatively higher with a reduction of commensals. Most of the shared Amplicon Sequencing Variants between cecum and blood in co-infected birds belonged to Pseudomonas, Staphylococcus, and members of Enterobacteriaceae while those assigned as Lactobacillus and members of Ruminococcaceae and Lachnospiraceae were found mainly in negative controls. In infected birds, E. coli in the cecal lumen penetrated into deeper layers, a phenomenon noticed with higher incidence in the dead and co-infected birds. Furthermore, numbers of lux-tagged E. coli in caeca were significantly higher at every sampling date in co-infected birds. Altogether, infection of layers with H. meleagridis and E. coli resulted in more severe pathological changes, dramatic shift in the cecal mucosa-associated microbiota, higher tissue colonization of pathogenic bacteria such as avian pathogenic E. coli in the gut and increased penetration of E. coli from the cecal lumen toward peritoneum. This study provides novel insights into the parasite-bacteria interaction in vivohighlighting the role of H. meleagridis to support E. coli in the pathogenesis of colibacillosis in chickens.
Using Colonization Assays and Comparative Genomics To Discover Symbiosis Behaviors and Factors in Vibrio fischeri.
2020 - mBio, 2: in press
Abstract:
The luminous marine Gram-negative bacterium () is the natural light organ symbiont of several squid species, including the Hawaiian bobtail squid, , and the Japanese bobtail squid, Work with has shown how the bacteria establish their niche in the light organ of the newly hatched host. Two types of strains have been distinguished based upon their behavior in cocolonization competition assays in juvenile , i.e., (i) niche-sharing or (ii) niche-dominant behavior. This study aimed to determine whether these behaviors are observed with other strains or whether they are specific to those isolated from light organs. Cocolonization competition assays between strains isolated from the congeneric squid or from other marine animals revealed the same sharing or dominant behaviors. In addition, whole-genome sequencing of these strains showed that the dominant behavior is polyphyletic and not associated with the presence or absence of a single gene or genes. Comparative genomics of 44 squid light organ isolates from around the globe led to the identification of symbiosis-specific candidates in the genomes of these strains. Colonization assays using genetic derivatives with deletions of these candidates established the importance of two such genes in colonization. This study has allowed us to expand the concept of distinct colonization behaviors to strains isolated from a number of squid and fish hosts. There is an increasing recognition of the importance of strain differences in the ecology of a symbiotic bacterial species and, in particular, how these differences underlie crucial interactions with their host. Nevertheless, little is known about the genetic bases for these differences, how they manifest themselves in specific behaviors, and their distribution among symbionts of different host species. In this study, we sequenced the genomes of isolated from the tissues of squids and fishes and applied comparative genomics approaches to look for patterns between symbiont lineages and host colonization behavior. In addition, we identified the only two genes that were exclusively present in all strains isolated from the light organs of sepiolid squid species. Mutational studies of these genes indicated that they both played a role in colonization of the squid light organ, emphasizing the value of applying a comparative genomics approach in the study of symbioses.
Environmental and intestinal phylum Firmicutes bacteria metabolize the plant sugar sulfoquinovose via a 6-deoxy-6-sulfofructose transaldolase pathway
2020 - iScience, 23: 101510
Abstract:
Bacterial degradation of the sugar sulfoquinovose (SQ, 6-deoxy-6-sulfoglucose) produced by plants, algae and cyanobacteria, is an important component of the biogeochemical carbon and sulfur cycles. Here, we reveal a third biochemical pathway for primary SQ degradation in an aerobic Bacillus aryabhattaistrain. An isomerase converts SQ to 6-deoxy-6-sulfofructose (SF). A novel transaldolase enzyme cleaves the SF to 3-sulfolactaldehyde (SLA), while the non-sulfonated C3-(glycerone)-moiety is transferred to an acceptor molecule, glyceraldehyde phosphate (GAP), yielding fructose-6-phosphate (F6P). Intestinal anaerobic bacteria such as Enterococcus gilvus, Clostridium symbiosum and Eubacterium rectale strains also express transaldolase-pathway gene clusters during fermentative growth with SQ. The now three known biochemical strategies for SQ catabolism reflect adaptations to the aerobic or anaerobic life-style of the different bacteria. The occurrence of these pathways in intestinal (family) Enterobacteriaceae and (phylum) Firmicutes strains further highlights a potential importance of metabolism of green-diet SQ by gut microbial communities to, ultimately, hydrogen sulfide.
Molecular causes of an evolutionary shift along the parasitism-mutualism continuum in a bacterial symbiont.
2020 - Proc. Natl. Acad. Sci. U.S.A., 117: 21658-21666
Abstract:
Symbiosis with microbes is a ubiquitous phenomenon with a massive impact on all living organisms, shaping the world around us today. Theoretical and experimental studies show that vertical transmission of symbionts leads to the evolution of mutualistic traits, whereas horizontal transmission facilitates the emergence of parasitic features. However, these studies focused on phenotypic data, and we know little about underlying molecular changes at the genomic level. Here, we combined an experimental evolution approach with infection assays, genome resequencing, and global gene expression analysis to study the effect of transmission mode on an obligate intracellular bacterial symbiont. We show that a dramatic shift in the frequency of genetic variants, coupled with major changes in gene expression, allow the symbiont to alter its position in the parasitism-mutualism continuum depending on the mode of between-host transmission. We found that increased parasitism in horizontally transmitted chlamydiae residing in amoebae was a result of processes occurring at the infectious stage of the symbiont's developmental cycle. Specifically, genes involved in energy production required for extracellular survival and the type III secretion system-the symbiont's primary virulence mechanism-were significantly up-regulated. Our results identify the genomic and transcriptional dynamics sufficient to favor parasitic or mutualistic strategies.
Chlamydiae in the Environment.
2020 - Trends Microbiol, 11: 877-888
Abstract:
Chlamydiae have been known for more than a century as major pathogens of humans. Yet they are also found ubiquitously in the environment where they thrive within protists and in an unmatched wide range of animals. This review summarizes recent advances in understanding chlamydial diversity and distribution in nature. Studying these environmental chlamydiae provides a novel perspective on basic chlamydial biology and evolution. A picture is beginning to emerge with chlamydiae representing one of the evolutionarily most ancient and successful groups of obligate intracellular bacteria.
Microbiome definition re-visited: old concepts and new challenges.
2020 - Microbiome, 1: 103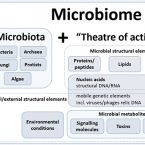
Abstract:
The field of microbiome research has evolved rapidly over the past few decades and has become a topic of great scientific and public interest. As a result of this rapid growth in interest covering different fields, we are lacking a clear commonly agreed definition of the term "microbiome." Moreover, a consensus on best practices in microbiome research is missing. Recently, a panel of international experts discussed the current gaps in the frame of the European-funded MicrobiomeSupport project. The meeting brought together about 40 leaders from diverse microbiome areas, while more than a hundred experts from all over the world took part in an online survey accompanying the workshop. This article excerpts the outcomes of the workshop and the corresponding online survey embedded in a short historical introduction and future outlook. We propose a definition of microbiome based on the compact, clear, and comprehensive description of the term provided by Whipps et al. in 1988, amended with a set of novel recommendations considering the latest technological developments and research findings. We clearly separate the terms microbiome and microbiota and provide a comprehensive discussion considering the composition of microbiota, the heterogeneity and dynamics of microbiomes in time and space, the stability and resilience of microbial networks, the definition of core microbiomes, and functionally relevant keystone species as well as co-evolutionary principles of microbe-host and inter-species interactions within the microbiome. These broad definitions together with the suggested unifying concepts will help to improve standardization of microbiome studies in the future, and could be the starting point for an integrated assessment of data resulting in a more rapid transfer of knowledge from basic science into practice. Furthermore, microbiome standards are important for solving new challenges associated with anthropogenic-driven changes in the field of planetary health, for which the understanding of microbiomes might play a key role. Video Abstract.
Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria).
2020 - ISME J, 1: 104-122
Abstract:
Most autotrophs use the Calvin-Benson-Bassham (CBB) cycle for carbon fixation. In contrast, all currently described autotrophs from the Campylobacterota (previously Epsilonproteobacteria) use the reductive tricarboxylic acid cycle (rTCA) instead. We discovered campylobacterotal epibionts ("Candidatus Thiobarba") of deep-sea mussels that have acquired a complete CBB cycle and may have lost most key genes of the rTCA cycle. Intriguingly, the phylogenies of campylobacterotal CBB cycle genes suggest they were acquired in multiple transfers from Gammaproteobacteria closely related to sulfur-oxidizing endosymbionts associated with the mussels, as well as from Betaproteobacteria. We hypothesize that "Ca. Thiobarba" switched from the rTCA cycle to a fully functional CBB cycle during its evolution, by acquiring genes from multiple sources, including co-occurring symbionts. We also found key CBB cycle genes in free-living Campylobacterota, suggesting that the CBB cycle may be more widespread in this phylum than previously known. Metatranscriptomics and metaproteomics confirmed high expression of CBB cycle genes in mussel-associated "Ca. Thiobarba". Direct stable isotope fingerprinting showed that "Ca. Thiobarba" has typical CBB signatures, suggesting that it uses this cycle for carbon fixation. Our discovery calls into question current assumptions about the distribution of carbon fixation pathways in microbial lineages, and the interpretation of stable isotope measurements in the environment.
Book chapters and other publications
STILLLEBEN with Symbionts
2020 - Performance Research, 25: 83-87
Abstract:
STILLLEBEN. Becoming Symbionts’ proposes to value milk and its microbial constituents as primordial assets and currencies - along with cells, sperm, blood, water, and oxygen. The latest scientific research, which has suggested that there is an intimate unseen interplay between mothers and their babies via the transfer of breast milk and microbes, which actually increases the value of the currency with each exchange. The co-operation of Irini Athanassakis and David Berry is an invitation to perceive the transfer of milk not only as an interaction visible to the naked eye, but also on the microscopic level of cells and bacteria. In order to challenge us with this unseen perspective, Athanassakis encourages us to step forward and take a performative and procreative role in expanding our perception. As we are home to billions of microscopic entities, we continuously cast this part of ourselves into our surroundings, impacting and interacting with everything around us. We leave a microbial trace, a lingering residue of cells on the objects, rooms, and people that we encounter. Breast milk and formula are part of such an exchange process. If we look at the logic of microbial exchange in nursing as ‘giving’ and knowledge about symbionts for a future holo-economy based on co-operation and mutualism for collective survival.
Draft genome sequences of Chlamydiales bacterium STE3 and Neochlamydia sp. AcF84, endosymbionts of Acanthamoeba spp.
2020 - Microbiol Resour Announc, 9: e00220-20
Abstract:
Chlamydiales bacterium STE3 and Neochlamydia sp. strain AcF84 are obligate intracellular symbionts of Acanthamoeba spp. isolated from the biofilm of a littoral cave wall and gills from striped tiger leaf fish, respectively. We report the draft genome sequences of these two environmental chlamydiae affiliated with the family Parachlamydiaceae.
Thinking outside the Chlamydia box
2020 - 429-458. in Chlamydia Biology. (M Tan, JH Hegemann, C Sütterlin). Caister Academic Press
Abstract:
Chlamydiae have long been studied exclusively in the context of disease. Yet, accumulating evidence over nearly three decades shows that chlamydiae are ubiquitous in the environment, thriving as symbionts of unicellular eukaryotes such as amoeba and infecting a broad range of animal hosts. These chlamydiae share the characteristic chlamydial developmental cycle and other chlamydial hallmarks. Their discovery fundamentally changed our perspective on chlamydial diversity. Instead of a single genus, Chlamydia, including closely related pathogens, the chlamydiae comprise hundreds of families and genera. Investigating isolates and non-cultured representatives provided insights into features that are in common with or divergent from known Chlamydia species, and suggested that some of these chlamydiae may also be considered pathogens. Importantly, these studies have contributed to a better understanding of the biology of all chlamydiae, and they provide a framework for investigating the evolution of the chlamydial intracellular lifestyle and pathogenicity.
Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at