Publications
Publications in peer reviewed journals
Organ transcriptomes of the lucinid clam Loripes orbiculatus (Poli, 1791) provide insights into their specialised roles in the biology of a chemosymbiotic bivalve.
2019 - BMC Genomics, 1: 820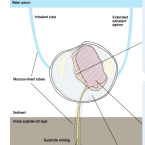
Abstract:
The lucinid clam Loripes orbiculatus lives in a nutritional symbiosis with sulphur-oxidizing bacteria housed in its gills. Although our understanding of the lucinid endosymbiont physiology and metabolism has made significant progress, relatively little is known about how the host regulates the symbiosis at the genetic and molecular levels. We generated transcriptomes from four L. orbiculatus organs (gills, foot, visceral mass, and mantle) for differential expression analyses, to better understand this clam's physiological adaptations to a chemosymbiotic lifestyle, and how it regulates nutritional and immune interactions with its symbionts.
The transcriptome profile of the symbiont-housing gill suggests the regulation of apoptosis and innate immunity are important processes in this organ. We also identified many transcripts encoding ion transporters from the solute carrier family that possibly allow metabolite exchange between host and symbiont. Despite the clam holobiont's clear reliance on chemosynthesis, the clam's visceral mass, which contains the digestive tract, is characterised by enzymes involved in digestion, carbohydrate recognition and metabolism, suggesting that L. orbiculatus has a mixotrophic diet. The foot transcriptome is dominated by the biosynthesis of glycoproteins for the construction of mucus tubes, and receptors that mediate the detection of chemical cues in the environment.
The transcriptome profiles of gills, mantle, foot and visceral mass provide insights into the molecular basis underlying the functional specialisation of bivalve organs adapted to a chemosymbiotic lifestyle.Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels.
2019 - Nat Microbiol, 12: 2487-2497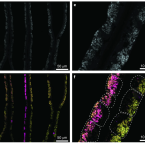
Abstract:
Genetic diversity of closely related free-living microorganisms is widespread and underpins ecosystem functioning, but most evolutionary theories predict that it destabilizes intimate mutualisms. Accordingly, strain diversity is assumed to be highly restricted in intracellular bacteria associated with animals. Here, we sequenced metagenomes and metatranscriptomes of 18 Bathymodiolus mussel individuals from four species, covering their known distribution range at deep-sea hydrothermal vents in the Atlantic. We show that as many as 16 strains of intracellular, sulfur-oxidizing symbionts coexist in individual Bathymodiolus mussels. Co-occurring symbiont strains differed extensively in key functions, such as the use of energy and nutrient sources, electron acceptors and viral defence mechanisms. Most strain-specific genes were expressed, highlighting their potential to affect fitness. We show that fine-scale diversity is pervasive in Bathymodiolus sulfur-oxidizing symbionts, and hypothesize that it may be widespread in low-cost symbioses where the environment, rather than the host, feeds the symbionts.
Summer phyto- and bacterioplankton communities during low and high productivity scenarios in the Western Antarctic Peninsula
2019 - Polar Biology, 42: 159-169
Abstract:
Phytoplankton blooms taking place during the warm season drive high productivity in Antarctic coastal seawaters. Important temporal and spatial variations exist in productivity patterns, indicating local constraints influencing the phototrophic community. Surface water in Chile Bay (Greenwich Island, South Shetlands) is influenced by freshwater from the melting of sea ice and surrounding glaciers; however, it is not a widely studied system. The phyto- and bacterioplankton communities in Chile Bay were studied over two consecutive summers; during a low productivity period (chlorophyll a < 0.05 mg m−3) and an ascendant phototrophic bloom (chlorophyll a up to 2.38 mg m−3). Microbial communities were analyzed by 16S rRNA—including plastidial—gene sequencing. Diatoms (mainly Thalassiosirales) were the most abundant phytoplankton, particularly during the ascendant bloom. Bacterioplankton in the low productivity period was less diverse and dominated by a few operational taxonomic units (OTUs), related to Colwellia and Pseudoalteromonas. Alpha diversity was higher during the bloom, where several Bacteroidetes taxa absent in the low productivity period were present. Network analysis indicated that phytoplankton relative abundance was correlated with bacterioplankton phylogenetic diversity and the abundance of several bacterial taxa. Hubs—the most connected OTUs in the network—were not the most abundant OTUs and included some poorly described taxa in Antarctica, such as Neptunomonas and Ekhidna. In summary, the results of this study indicate that in Antarctic Peninsula coastal waters, such as Chile Bay, higher bacterioplankton community diversity occurs during a phototrophic bloom. This is likely a result of primary production, providing a source of fresh organic matter to bacterioplankton.
Bacterial community structure in a sympagic habitat expanding with global warming: brackish ice brine at 85-90 °N.
2019 - ISME J, 2: 316-333
Abstract:
Larger volumes of sea ice have been thawing in the Central Arctic Ocean (CAO) during the last decades than during the past 800,000 years. Brackish brine (fed by meltwater inside the ice) is an expanding sympagic habitat in summer all over the CAO. We report for the first time the structure of bacterial communities in this brine. They are composed of psychrophilic extremophiles, many of them related to phylotypes known from Arctic and Antarctic regions. Community structure displayed strong habitat segregation between brackish ice brine (IB; salinity 2.4-9.6) and immediate sub-ice seawater (SW; salinity 33.3-34.9), expressed at all taxonomic levels (class to genus), by dominant phylotypes as well as by the rare biosphere, and with specialists dominating IB and generalists SW. The dominant phylotypes in IB were related to Candidatus Aquiluna and Flavobacterium, those in SW to Balneatrix and ZD0405, and those shared between the habitats to Halomonas, Polaribacter and Shewanella. A meta-analysis for the oligotrophic CAO showed a pattern with Flavobacteriia dominating in melt ponds, Flavobacteriia and Gammaproteobacteria in solid ice cores, Flavobacteriia, Gamma- and Betaproteobacteria, and Actinobacteria in brine, and Alphaproteobacteria in SW. Based on our results, we expect that the roles of Actinobacteria and Betaproteobacteria in the CAO will increase with global warming owing to the increased production of meltwater in summer. IB contained three times more phylotypes than SW and may act as an insurance reservoir for bacterial diversity that can act as a recruitment base when environmental conditions change.
Polyclonal symbiont populations in hydrothermal vent tubeworms and the environment.
2019 - Proc. Biol. Sci., 1896: 20181281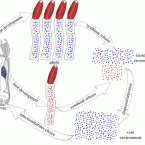
Abstract:
Horizontally transmitted symbioses usually house multiple and variable symbiont genotypes that are acquired from a much more diverse environmental pool via partner choice mechanisms. However, in the deep-sea hydrothermal vent tubeworm Riftia pachyptila (Vestimentifera, Siboglinidae), it has been suggested that the Candidatus Endoriftia persephone symbiont is monoclonal. Here, we show with high-coverage metagenomics that adult R. pachyptila house a polyclonal symbiont population consisting of one dominant and several low-frequency variants. This dominance of one genotype is confirmed by multilocus gene sequencing of amplified housekeeping genes in a broad range of host individuals where three out of four loci ( atpA, uvrD and recA) revealed no genomic differences, while one locus ( gyrB) was more diverse in adults than in juveniles. We also analysed a metagenome of free-living Endoriftia and found that the free-living population showed greater sequence variability than the host-associated population. Most juveniles and adults shared a specific dominant genotype, while other genotypes can dominate in few individuals. We suggest that although generally permissive, partner choice is selective enough to restrict uptake of some genotypes present in the environment.
Genomic Features for Desiccation Tolerance and Sugar Biosynthesis in the Extremophile sp. UTEX B3054.
2019 - Front Microbiol, 950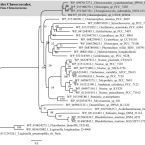
Abstract:
For tolerating extreme desiccation, cyanobacteria are known to produce both compatible solutes at intracellular level and a copious amount of exopolysaccharides as a protective coat. However, these molecules make cyanobacterial cells refractory to a broad spectrum of cell disruption methods, hindering genome sequencing, and molecular studies. In fact, few genomes are already available from cyanobacteria from extremely desiccated environments such as deserts. In this work, we report the 5.4 Mbp draft genome (with 100% of completeness in 105 contigs) of sp. UTEX B3054 (subsection I; Order Chroococcales), a cultivable sugar-rich and hardly breakable hypolithic cyanobacterium from the Atacama Desert. Our analyses focused on genomic features related to sugar-biosynthesis and adaptation to dryness. Among other findings, screening of genome revealed a unique genetic potential related to the biosynthesis and regulation of compatible solutes and polysaccharides. For instance, our findings showed for the first time a novel genomic arrangement exclusive of Chroococcaceae cyanobacteria associated with the recycling of trehalose, a compatible solute involved in desiccation tolerance. Additionally, we performed a comparative genome survey and analyses to entirely predict the highly diverse pool of glycosyltransferases enzymes, key players in polysaccharide biosynthesis and the formation of a protective coat to dryness. We expect that this work will set the fundamental genomic framework for further research on microbial tolerance to desiccation and to a wide range of other extreme environmental conditions. The study of microorganisms like sp. UTEX B3054 will contribute to expand our limited understanding regarding water optimization and molecular mechanisms allowing extremophiles to thrive in xeric environments such as the Atacama Desert.
Interactions in self-assembled microbial communities saturate with diversity.
2019 - ISME J, 6: 1602-1617
Abstract:
How the diversity of organisms competing for or sharing resources influences community function is an important question in ecology but has rarely been explored in natural microbial communities. These generally contain large numbers of species making it difficult to disentangle how the effects of different interactions scale with diversity. Here, we show that changing diversity affects measures of community function in relatively simple communities but that increasing richness beyond a threshold has little detectable effect. We generated self-assembled communities with a wide range of diversity by growth of cells from serially diluted seawater on brown algal leachate. We subsequently isolated the most abundant taxa from these communities via dilution-to-extinction in order to compare productivity functions of the entire community to those of individual taxa. To parse the effect of different types of organismal interactions, we defined relative total function (RTF) as an index for positive or negative effects of diversity on community function. Our analysis identified three overall regimes with increasing diversity. At low richness (<12 taxa), positive and negative effects of interactions were both weak, while at moderate richness (12-26 taxa), community resource uptake increased but the carbon use efficiency decreased. Finally, beyond 26 taxa, the effect of interactions on community function saturated and further diversity increases did not affect community function. Although more diverse communities had overall greater access to resources, on average individual taxa within these communities had lower resource availability and reduced carbon use efficiency. Our results thus suggest competition and complementation simultaneously increase with diversity but both saturate at a threshold.
Maintenance of Sympatric and Allopatric Populations in Free-Living Terrestrial Bacteria.
2019 - mBio, 5: e02361-19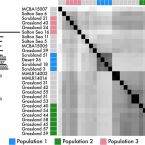
Abstract:
For free-living bacteria and archaea, the equivalent of the biological species concept does not exist, creating several obstacles to the study of the processes contributing to microbial diversification. These obstacles are particularly high in soil, where high bacterial diversity inhibits the study of closely related genotypes and therefore the factors structuring microbial populations. Here, we isolated strains within a single ecotype from surface soil (leaf litter) across a regional climate gradient and investigated the phylogenetic structure, recombination, and flexible gene content of this genomic diversity to infer patterns of gene flow. Our results indicate that microbial populations are delineated by gene flow discontinuities, with distinct populations cooccurring at multiple sites. Bacterial population structure was further delineated by genomic features allowing for the identification of candidate genes possibly contributing to local adaptation. These results suggest that the genetic structure within this bacterium is maintained both by ecological specialization in localized microenvironments (isolation by environment) and by dispersal limitation between geographic locations (isolation by distance). Due to the promiscuous exchange of genetic material and asexual reproduction, delineating microbial species (and, by extension, populations) remains challenging. Because of this, the vast majority of microbial studies assessing population structure often compare divergent strains from disparate environments under varied selective pressures. Here, we investigated the population structure within a single bacterial ecotype, a unit equivalent to a eukaryotic species, defined as highly clustered genotypic and phenotypic strains with the same ecological niche. Using a combination of genomic and computational analyses, we assessed the phylogenetic structure, extent of recombination, and flexible gene content of this genomic diversity to infer patterns of gene flow. To our knowledge, this study is the first to do so for a dominant soil bacterium. Our results indicate that bacterial soil populations, similarly to those in other environments, are structured by gene flow discontinuities and exhibit distributional patterns consistent with both isolation by distance and isolation by environment. Thus, both dispersal limitation and local environments contribute to the divergence among closely related soil bacteria as observed in macroorganisms.
A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations.
2019 - Cell, 4: 820-834.e14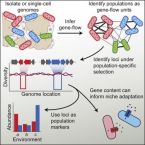
Abstract:
Delineating ecologically meaningful populations among microbes is important for identifying their roles in environmental and host-associated microbiomes. Here, we introduce a metric of recent gene flow, which when applied to co-existing microbes, identifies congruent genetic and ecological units separated by strong gene flow discontinuities from their next of kin. We then develop a pipeline to identify genome regions within these units that show differential adaptation and allow mapping of populations onto environmental variables or host associations. Using this reverse ecology approach, we show that the human commensal bacterium Ruminococcus gnavus breaks up into sharply delineated populations that show different associations with health and disease. Defining populations by recent gene flow in this way will facilitate the analysis of bacterial and archaeal genomes using ecological and evolutionary theory developed for plants and animals, thus allowing for testing unifying principles across all biology.
A Bioinformatics Guide to Plant Microbiome Analysis.
2019 - Front Plant Sci, 1313
Abstract:
Recent evidence for intimate relationship of plants with their microbiota shows that plants host individual and diverse microbial communities that are essential for their survival. Understanding their relatedness using genome-based and high-throughput techniques remains a hot topic in microbiome research. Molecular analysis of the plant holobiont necessitates the application of specific sampling and preparatory steps that also consider sources of unwanted information, such as soil, co-amplified plant organelles, human DNA, and other contaminations. Here, we review state-of-the-art and present practical guidelines regarding experimental and computational aspects to be considered in molecular plant-microbiome studies. We discuss sequencing and "omics" techniques with a focus on the requirements needed to adapt these methods to individual research approaches. The choice of primers and sequence databases is of utmost importance for amplicon sequencing, while the assembly and binning of shotgun metagenomic sequences is crucial to obtain quality data. We discuss specific bioinformatic workflows to overcome the limitation of genome database resources and for covering large eukaryotic genomes such as fungi. In transcriptomics, it is necessary to account for the separation of host mRNA or dual-RNAseq data. Metaproteomics approaches provide a snapshot of the protein abundances within a plant tissue which requires the knowledge of complete and well-annotated plant genomes, as well as microbial genomes. Metabolomics offers a powerful tool to detect and quantify small molecules and molecular changes at the plant-bacteria interface if the necessary requirements with regard to (secondary) metabolite databases are considered. We highlight data integration and complementarity which should help to widen our understanding of the interactions among individual players of the plant holobiont in the future.
Hair eruption initiates and commensal skin microbiota aggravate adverse events of anti-EGFR therapy
2019 - Sci Transl Med, 11: eaax2693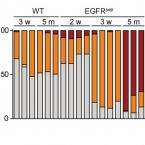
Abstract:
Epidermal growth factor receptor (EGFR)–targeted anticancer therapy induces stigmatizing skin toxicities affecting patients’ quality of life and therapy adherence. The lack of mechanistic details underlying these adverse events hampers their management. We found that EGFR/ERK signaling is required in LRIG1-positive stem cells during de novo hair eruption to secure barrier integrity and prevent the invasion of commensal microbiota and inflammatory skin disease. EGFR-deficient epidermis is permissive for microbiota outgrowth and displays an atopic-like TH2-dominated signature. The opening of the follicular ostia during hair eruption allows invasion of commensal microbiota into the hair follicle, initiating an additional TH1 and TH17 response culminating in chronic folliculitis. Restoration of epidermal ERK signaling via prophylactic FGF7 treatment or transgenic SOS expression rescues the barrier defect in the absence of EGFR, highlighting a therapeutic anchor point. These data reveal that commensal skin microbiota provoke atopic-like inflammatory skin diseases by invading into the follicular opening of erupting hair.
Berry-Enriched Diet in Salt-Sensitive Hypertensive Rats: Metabolic Fate of (Poly)Phenols and the Role of Gut Microbiota.
2019 - Nutrients, 11: 2634
Abstract:
Diets rich in (poly)phenols are associated with a reduced reduction in the incidence of cardiovascular disorders. While the absorption and metabolism of (poly)phenols has been described, it is not clear how their metabolic fate is affected under pathological conditions. This study evaluated the metabolic fate of berry (poly)phenols in an in vivo model of hypertension as well as the associated microbiota response. Dahl salt-sensitive rats were fed either a low-salt diet (0.26% NaCl) or a high-salt diet (8% NaCl), with or without a berry mixture (blueberries, blackberries, raspberries, Portuguese crowberry and strawberry tree fruit) for 9 weeks. The salt-enriched diet promoted an increase in the urinary excretion of berry (poly)phenol metabolites, while the abundance of these metabolites decreased in faeces, as revealed by UPLC-MS/MS. Moreover, salt and berries modulated gut microbiota composition as demonstrated by 16S rRNA analysis. Some changes in the microbiota composition were associated with the high-salt diet and revealed an expansion of the families and . However, this effect was mitigated by the dietary supplementation with berries. Alterations in the metabolic fate of (poly)phenols occur in parallel with the modulation of gut microbiota in hypertensive rats. Thus, beneficial effects of (poly)phenols could be related with these interlinked modifications, between metabolites and microbiota environments.
Glacial runoff promotes deep burial of sulfur cycling-associated microorganisms in marine sediments
2019 - Front Microbiol, 10: 2558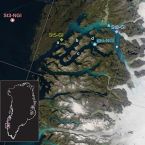
Abstract:
Marine fjords with active glacier outlets are hot spots for organic matter burial in the sediments and subsequent microbial mineralization. Here, we investigated controls on microbial community assembly in sub-arctic glacier-influenced (GI) and non-glacier-influenced (NGI) marine sediments in the Godthåbsfjord region, south-western Greenland. We used a correlative approach integrating 16S rRNA gene and dissimilatory sulfite reductase (dsrB) amplicon sequence data over six meters of depth with biogeochemistry, sulfur-cycling activities, and sediment ages. GI sediments were characterized by comparably high sedimentation rates and had ‘young’ sediment ages of <500 years even at 6 m sediment depth. In contrast, NGI stations reached ages of approximately 10,000 years at these depths. Sediment age-depth relationships, sulfate reduction rates, and C/N ratios were strongly correlated with differences in microbial community composition between GI and NGI sediments, indicating that age and diagenetic state were key drivers of microbial community assembly in subsurface sediments. Similar bacterial and archaeal communities were present in the surface sediments of all stations, whereas only in GI sediments were many surface taxa also abundant through the whole sediment core. The relative abundance of these taxa, including diverse Desulfobacteraceae members, correlated positively with sulfate reduction rates, indicating their active contributions to sulfur-cycling processes. In contrast, other surface community members, such as Desulfatiglans, Atribacteria and Chloroflexi, survived the slow sediment burial at NGI stations and dominated in the deepest sediment layers. These taxa are typical for the energy-limited marine deep biosphere and their relative abundances correlated positively with sediment age. In conclusion, our data suggests that high rates of sediment accumulation caused by glacier runoff and associated changes in biogeochemistry, promote persistence of sulfur-cycling activity and burial of a larger fraction of the surface microbial community into the deep subsurface.
In situ abundance and carbon fixation activity of distinct anoxygenic phototrophs in the stratified seawater lake Rogoznica.
2019 - Environ. Microbiol., 10: 3896-3908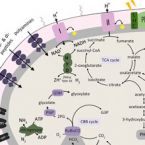
Abstract:
Sulphide-driven anoxygenic photosynthesis is an ancient microbial metabolism that contributes significantly to inorganic carbon fixation in stratified, sulphidic water bodies. Methods commonly applied to quantify inorganic carbon fixation by anoxygenic phototrophs, however, cannot resolve the contributions of distinct microbial populations to the overall process. We implemented a straightforward workflow, consisting of radioisotope labelling and flow cytometric cell sorting based on the distinct autofluorescence of bacterial photopigments, to discriminate and quantify contributions of co-occurring anoxygenic phototrophic populations to in situ inorganic carbon fixation in environmental samples. This allowed us to assign 89.3% ± 7.6% of daytime inorganic carbon fixation by anoxygenic phototrophs in Lake Rogoznica (Croatia) to an abundant chemocline-dwelling population of green sulphur bacteria (dominated by Chlorobium phaeobacteroides), whereas the co-occurring purple sulphur bacteria (Halochromatium sp.) contributed only 1.8% ± 1.4%. Furthermore, we obtained two metagenome assembled genomes of green sulphur bacteria and one of a purple sulphur bacterium which provides the first genomic insights into the genus Halochromatium, confirming its high metabolic flexibility and physiological potential for mixo- and heterotrophic growth.
A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome.
2019 - Nat Commun, 1: 4366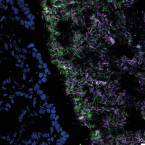
Abstract:
Compartmentalization of the gut microbiota is thought to be important to system function, but the extent of spatial organization in the gut ecosystem remains poorly understood. Here, we profile the murine colonic microbiota along longitudinal and lateral axes using laser capture microdissection. We found fine-scale spatial structuring of the microbiota marked by gradients in composition and diversity along the length of the colon. Privation of fiber reduces the diversity of the microbiota and disrupts longitudinal and lateral gradients in microbiota composition. Both mucus-adjacent and luminal communities are influenced by the absence of dietary fiber, with the loss of a characteristic distal colon microbiota and a reduction in the mucosa-adjacent community, concomitant with depletion of the mucus layer. These results indicate that diet has not only global but also local effects on the composition of the gut microbiota, which may affect function and resilience differently depending on location.
Horizontally transmitted symbiont populations in deep-sea mussels are genetically isolated.
2019 - ISME J, 12: 2954-2968
Abstract:
Eukaryotes are habitats for bacterial organisms where the host colonization and dispersal among individual hosts have consequences for the bacterial ecology and evolution. Vertical symbiont transmission leads to geographic isolation of the microbial population and consequently to genetic isolation of microbiotas from individual hosts. In contrast, the extent of geographic and genetic isolation of horizontally transmitted microbiota is poorly characterized. Here we show that chemosynthetic symbionts of individual Bathymodiolus brooksi mussels constitute genetically isolated subpopulations. The reconstruction of core genome-wide strains from high-resolution metagenomes revealed distinct phylogenetic clades. Nucleotide diversity and strain composition vary along the mussel life span and individual hosts show a high degree of genetic isolation. Our results suggest that the uptake of environmental bacteria is a restricted process in B. brooksi, where self-infection of the gill tissue results in serial founder effects during symbiont evolution. We conclude that bacterial colonization dynamics over the host life cycle is thus an important determinant of population structure and genome evolution of horizontally transmitted symbionts.
Chemosymbiotic bivalves contribute to the nitrogen budget of seagrass ecosystems.
2019 - ISME J, 12: 3131-3134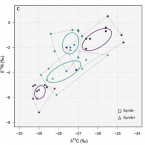
Abstract:
In many seagrass sediments, lucinid bivalves and their sulfur-oxidizing symbionts are thought to underpin key ecosystem functions, but little is known about their role in nutrient cycles, particularly nitrogen. We used natural stable isotopes, elemental analyses, and stable isotope probing to study the ecological stoichiometry of a lucinid symbiosis in spring and fall. Chemoautotrophy appeared to dominate in fall, when chemoautotrophic carbon fixation rates were up to one order of magnitude higher as compared with the spring, suggesting a flexible nutritional mutualism. In fall, an isotope pool dilution experiment revealed carbon limitation of the symbiosis and ammonium excretion rates up to tenfold higher compared with fluxes reported for nonsymbiotic marine bivalves. These results provide evidence that lucinid bivalves can contribute substantial amounts of ammonium to the ecosystem. Given the preference of seagrasses for this nitrogen source, lucinid bivalves' contribution may boost productivity of these important blue carbon ecosystems.
On the evolution and physiology of cable bacteria.
2019 - Proc. Natl. Acad. Sci. U.S.A., 38: 19116-19125
Abstract:
Cable bacteria of the family Desulfobulbaceae form centimeter-long filaments comprising thousands of cells. They occur worldwide in the surface of aquatic sediments, where they connect sulfide oxidation with oxygen or nitrate reduction via long-distance electron transport. In the absence of pure cultures, we used single-filament genomics and metagenomics to retrieve draft genomes of 3 marine Electrothrix and 1 freshwater Electronema species. These genomes contain >50% unknown genes but still share their core genomic makeup with sulfate-reducing and sulfur-disproportionating Desulfobulbaceae, with few core genes lost and 212 unique genes (from 197 gene families) conserved among cable bacteria. Last common ancestor analysis indicates gene divergence and lateral gene transfer as equally important origins of these unique genes. With support from metaproteomics of a Electronema enrichment, the genomes suggest that cable bacteria oxidize sulfide by reversing the canonical sulfate reduction pathway and fix CO using the Wood-Ljungdahl pathway. Cable bacteria show limited organotrophic potential, may assimilate smaller organic acids and alcohols, fix N, and synthesize polyphosphates and polyglucose as storage compounds; several of these traits were confirmed by cell-level experimental analyses. We propose a model for electron flow from sulfide to oxygen that involves periplasmic cytochromes, yet-unidentified conductive periplasmic fibers, and periplasmic oxygen reduction. This model proposes that an active cable bacterium gains energy in the anodic, sulfide-oxidizing cells, whereas cells in the oxic zone flare off electrons through intense cathodic oxygen respiration without energy conservation; this peculiar form of multicellularity seems unparalleled in the microbial world.
Antioxidative activity and health benefits of anthocyanin-rich fruit juice in healthy volunteers.
2019 - Free Radic. Res., 1-11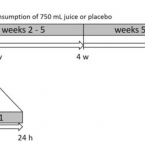
Abstract:
Oxidative cell damage has been linked to the pathogenesis of numerous diseases such as atherosclerosis, type 2 diabetes, and cancer. The consumption of foods rich in polyphenols (e.g. anthocyanins) has been shown to exert preventive effects against such diseases. We investigated the biological effects of anthocyanin-rich fruit juice in a 9-week, placebo-controlled intervention study with 57 healthy male volunteers. The study design encompassed an initial 1 week of wash-out, followed by 8 weeks of intervention period with anthocyanin-rich fruit juice or placebo. The anthocyanin-rich fruit juice demonstrated DNA-protective and antioxidant effects; however, the placebo beverage, rich in vitamin C, showed similar effects based on the tested biomarkers. A significant reduction in background and total DNA strand breaks was observed in both groups within 24 h as well as after 8 weeks of intervention. Only anthocyanin-rich fruit juice consumption provided a significant reduction in body fat and an increase in fat-free mass. The activity of superoxide dismutase (SOD) was significantly elevated after consumption of anthocyanin-rich fruit juice. Both groups showed decreased levels of LDL and total cholesterol (TC) within the first week of the intervention. Similar results in both groups could be explained by the relatively high vitamin C contents of both beverages (>500 mg/L), which may have masked the effects of anthocyanins and other antioxidants in the studied juice. Taken together, anthocyanin-rich fruit juice as well as the placebo drink, both of which had high vitamin C content, can improve DNA integrity and might influence lipid metabolism in humans.
Membrane Lipid Composition of the Moderately Thermophilic Ammonia-Oxidizing Archaeon " Nitrosotenuis uzonensis" at Different Growth Temperatures.
2019 - Appl. Environ. Microbiol., 20: 1–17
Abstract:
" Nitrosotenuis uzonensis" is the only cultured moderately thermophilic member of the thaumarchaeotal order (NP) that contains many mesophilic marine strains. We examined its membrane lipid composition at different growth temperatures (37°C, 46°C, and 50°C). Its lipids were all membrane-spanning glycerol dialkyl glycerol tetraethers (GDGTs), with 0 to 4 cyclopentane moieties. Crenarchaeol (cren), the characteristic thaumarchaeotal GDGT, and its isomer (cren') were present in high abundance (30 to 70%). The GDGT polar headgroups were mono-, di-, and trihexoses and hexose/phosphohexose. The ratio of glycolipid to phospholipid GDGTs was highest in the cultures grown at 50°C. With increasing growth temperatures, the relative contributions of cren and cren' increased, while those of GDGT-0 to GDGT-4 (including isomers) decreased. TEX (tetraether index of tetraethers consisting of 86 carbons)-derived temperatures were much lower than the actual growth temperatures, further demonstrating that TEX does not accurately reflect the membrane lipid adaptation of thermophilic As the temperature increased, specific GDGTs changed relative to their isomers, possibly representing temperature adaption-induced changes in cyclopentane ring stereochemistry. Comparison of a wide range of thaumarchaeotal core lipid compositions revealed that the " Nitrosotenuis uzonensis" cultures clustered separately from other members of the NP order and the (NS) order. While phylogeny generally seems to have a strong influence on GDGT distribution, our analysis of " Nitrosotenuis uzonensis" demonstrates that its terrestrial, higher-temperature niche has led to a lipid composition that clearly differentiates it from other NP members and that this difference is mostly driven by its high cren' content. For , the ratio of their glycerol dialkyl glycerol tetraether (GDGT) lipids depends on growth temperature, a premise that forms the basis of the widely applied TEX paleotemperature proxy. A thorough understanding of which GDGTs are produced by which and what the effect of temperature is on their GDGT composition is essential for constraining the TEX proxy. " Nitrosotenuis uzonensis" is a moderately thermophilic thaumarchaeote enriched from a thermal spring, setting it apart in its environmental niche from the other marine mesophilic members of its order. Indeed, we found that the GDGT composition of " Nitrosotenuis uzonensis" cultures was distinct from those of other members of its order and was more similar to those of other thermophilic, terrestrial This suggests that while phylogeny has a strong influence on GDGT distribution, the environmental niche that a thaumarchaeote inhabits also shapes its GDGT composition.
Expansion of Thaumarchaeota habitat range is correlated with horizontal transfer of ATPase operons.
2019 - ISME J, 12: 3067-3079
Abstract:
Thaumarchaeota are responsible for a significant fraction of ammonia oxidation in the oceans and in soils that range from alkaline to acidic. However, the adaptive mechanisms underpinning their habitat expansion remain poorly understood. Here we show that expansion into acidic soils and the high pressures of the hadopelagic zone of the oceans is tightly linked to the acquisition of a variant of the energy-yielding ATPases via horizontal transfer. Whereas the ATPase genealogy of neutrophilic Thaumarchaeota is congruent with their organismal genealogy inferred from concatenated conserved proteins, a common clade of V-type ATPases unites phylogenetically distinct clades of acidophilic/acid-tolerant and piezophilic/piezotolerant species. A presumptive function of pumping cytoplasmic protons at low pH is consistent with the experimentally observed increased expression of the V-ATPase in an acid-tolerant thaumarchaeote at low pH. Consistently, heterologous expression of the thaumarchaeotal V-ATPase significantly increased the growth rate of E. coli at low pH. Its adaptive significance to growth in ocean trenches may relate to pressure-related changes in membrane structure in which this complex molecular machine must function. Together, our findings reveal that the habitat expansion of Thaumarchaeota is tightly correlated with extensive horizontal transfer of atp operons.
Draft Genome Sequence of Desulfosporosinus fructosivorans Strain 63.6F, Isolated from Marine Sediment in the Baltic Sea
2019 - Microbiology Resource Announcements, 8: e00427-1
Abstract:
Desulfosporosinus fructosivorans strain 63.6FT is a strictly anaerobic, spore-forming, sulfate-reducing bacterium isolated from marine sediment in the Baltic Sea. Here, we report the draft genome sequence of D. fructosivorans 63.6FT.
Specific micropollutant biotransformation pattern by the comammox bacterium Nitrospira inopinata
2019 - Environ. Sci. Technol., 15: 8695-8705
Abstract:
The recently discovered complete ammonia-oxidizing (comammox) bacteria occur in various environments, including wastewater treatment plants. To better understand their role in micropollutant biotransformation in comparison with ammonia-oxidizing bacteria (AOB) and ammonia-oxidizing archaea (AOA), we investigated the biotransformation capability of (the only comammox isolate) for 17 micropollutants. Asulam, fenhexamid, mianserin, and ranitidine were biotransformed by , (AOA), and Nm90 (AOB). More distinctively, carbendazim, a benzimidazole fungicide, was exclusively biotransformed by . The biotransformation of carbendazim only occurred when was supplied with ammonia but not nitrite as the energy source. The exclusive biotransformation of carbendazim by was likely enabled by an enhanced substrate promiscuity of its unique AMO and its much higher substrate (for ammonia) affinity compared with the other two ammonia oxidizers. One major plausible transformation product (TP) of carbendazim is a hydroxylated form at the aromatic ring, which is consistent with the function of AMO. These findings provide fundamental knowledge on the micropollutant degradation potential of a comammox bacterium to better understand the fate of micropollutants in nitrifying environments.
Soil multifunctionality is affected by the soil environment and by microbial community composition and diversity.
2019 - Soil Biol. Biochem., 107521
Abstract:
Microorganisms are critical in mediating carbon (C) and nitrogen (N) cycling processes in soils. Yet, it has long been debated whether the processes underlying biogeochemical cycles are affected by the composition and diversity of the soil microbial community or not. The composition and diversity of soil microbial communities can be influenced by various environmental factors, which in turn are known to impact biogeochemical processes. The objectives of this study were to test effects of multiple edaphic drivers individually and represented as the multivariate soil environment interacting with microbial community composition and diversity, and concomitantly on multiple soil functions (i.e. soil enzyme activities, soil C and N processes). We employed high-throughput sequencing (Illumina MiSeq) to analyze bacterial/archaeal and fungal community composition by targeting the 16S rRNA gene and the ITS1 region of soils collected from three land uses (cropland, grassland and forest) deriving from two bedrock forms (silicate and limestone). Based on this data set we explored single and combined effects of edaphic variables on soil microbial community structure and diversity, as well as on soil enzyme activities and several soil C and N processes. We found that both bacterial/archaeal and fungal communities were shaped by the same edaphic factors, with most single edaphic variables and the combined soil environment representation exerting stronger effects on bacterial/archaeal communities than on fungal communities, as demonstrated by (partial) Mantel tests. We also found similar edaphic controls on the bacterial/archaeal/fungal richness and diversity. Soil C processes were only directly affected by the soil environment but not affected by microbial community composition. In contrast, soil N processes were significantly related to bacterial/archaeal community composition and bacterial/archaeal/fungal richness/diversity but not directly affected by the soil environment. This indicates direct control of the soil environment on soil C processes and indirect control of the soil environment on soil N processes by structuring the microbial communities. The study further highlights the importance of edaphic drivers and microbial communities (i.e. composition and diversity) on important soil C and N processes.
Diversity decoupled from sulfur isotope fractionation in a sulfate reducing microbial community
2019 - Geobiology, 17: 660-67
Abstract:
The extent of fractionation of sulfur isotopes by sulfate-reducing microbes is dictated by genomic and environmental factors. A greater understanding of species-specific fractionations may better inform interpretation of sulfur isotopes preserved in the rock record. To examine whether gene diversity influences net isotopic fractionation in situ, we assessed environmental chemistry, sulfate reduction rates, diversity of putative sulfur-metabolizing organisms by 16S rRNA and dissimilatory sulfite reductase (dsrB) gene amplicon sequencing, and net fractionation of sulfur isotopes along a sediment transect of a hypersaline Arctic spring. In situ sulfate reduction rates yielded minimum cell-specific sulfate reduction rates < 0.3 × 10-15 moles cell-1 day-1 . Neither 16S rRNA nor dsrB diversity indices correlated with relatively constant (38‰-45‰) net isotope fractionation (ε34 Ssulfide-sulfate ). Measured ε34 S values could be reproduced in a mechanistic fractionation model if 1%-2% of the microbial community (10%-60% of Deltaproteobacteria) were engaged in sulfate respiration, indicating heterogeneous respiratory activity within sulfate-reducing populations. This model indicated enzymatic kinetic diversity of Apr was more likely to correlate with sulfur fractionation than DsrB. We propose that, above a threshold Shannon diversity value of 0.8 for dsrB, the influence of the specific composition of the microbial community responsible for generating an isotope signal is overprinted by the control exerted by environmental variables on microbial physiology.
Draft Genome Sequence of Desulfosporosinus sp. Strain Sb-LF, Isolated from an acidic peatland in Germany
2019 - Microbiology Resource Announcements, 8: e00428-1
Abstract:
Desulfosporosinus sp. strain Sb-LF was isolated from an acidic peatland in Bavaria, Germany. Here, we report the draft genome sequence of the sulfate-reducing and lactate-utilizing strain Sb-LF.
Proteomic Response of Three Marine Ammonia-Oxidizing Archaea to Hydrogen Peroxide and Their Metabolic Interactions with a Heterotrophic Alphaproteobacterium.
2019 - mSystems, 4: e00181–e00119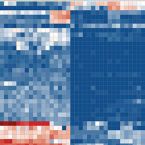
Abstract:
Ammonia-oxidizing archaea (AOA) play an important role in the nitrogen cycle and account for a considerable fraction of the prokaryotic plankton in the ocean. Most AOA lack the hydrogen peroxide (HO)-detoxifying enzyme catalase, and some AOA have been shown to grow poorly under conditions of exposure to HO However, differences in the degrees of HO sensitivity of different AOA strains, the physiological status of AOA cells exposed to HO, and their molecular response to HO remain poorly characterized. Further, AOA might rely on heterotrophic bacteria to detoxify HO, and yet the extent and variety of costs and benefits involved in these interactions remain unclear. Here, we used a proteomics approach to compare the protein profiles of three strains grown in the presence and absence of catalase and in coculture with the heterotrophic alphaproteobacterium We observed that most proteins detected at a higher relative abundance in HO-exposed cells had no known function in oxidative stress defense. Instead, these proteins were putatively involved in the remodeling of the extracellular matrix, which we hypothesize to be a strategy limiting the influx of HO into the cells. Using RNA-stable isotope probing, we confirmed that cells growing in coculture with the strains assimilated -derived organic carbon, suggesting that AOA could recruit HO-detoxifying bacteria through the release of labile organic matter. Our results contribute new insights into the response of AOA to HO and highlight the potential ecological importance of their interactions with heterotrophic free-living bacteria in marine environments. Ammonia-oxidizing archaea (AOA) are the most abundant chemolithoautotrophic microorganisms in the oxygenated water column of the global ocean. Although HO appears to be a universal by-product of aerobic metabolism, genes encoding the hydrogen peroxide (HO)-detoxifying enzyme catalase are largely absent in genomes of marine AOA. Here, we provide evidence that closely related marine AOA have different degrees of sensitivity to HO, which may contribute to niche differentiation between these organisms. Furthermore, our results suggest that marine AOA rely on HO detoxification during periods of high metabolic activity and release organic compounds, thereby potentially attracting heterotrophic prokaryotes that provide this missing function. In summary, this report provides insights into the metabolic interactions between AOA and heterotrophic bacteria in marine environments and suggests that AOA play an important role in the biogeochemical carbon cycle by making organic carbon available for heterotrophic microorganisms.
A Multicolor Fluorescence Hybridization Approach Using an Extended Set of Fluorophores to Visualize Microorganisms.
2019 - Front Microbiol, 10: 1383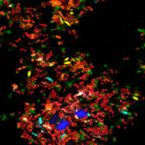
Abstract:
Fluorescence hybridization (FISH) with rRNA-targeted oligonucleotide probes is a key method for the detection of (uncultured) microorganisms in environmental and medical samples. A major limitation of standard FISH protocols, however, is the small number of phylogenetically distinct target organisms that can be detected simultaneously. In this study, we introduce a multicolor FISH approach that uses eight fluorophores with distinct spectral properties, which can unambiguously be distinguished by confocal laser scanning microscopy combined with white light laser technology. Hybridization of rRNA-targeted DNA oligonucleotide probes, which were mono-labeled with these fluorophores, to cultures confirmed that the fluorophores did not affect probe melting behavior. Application of the new multicolor FISH method enabled the differentiation of seven (potentially up to eight) phylogenetically distinct microbial populations in an artificial community of mixed pure cultures (five bacteria, one archaeon, and one yeast strain) and in activated sludge from a full-scale wastewater treatment plant. In contrast to previously published multicolor FISH approaches, this method does not rely on combinatorial labeling of the same microorganisms with different fluorophores, which is prone to biases. Furthermore, images acquired by this method do not require elaborate post-processing prior to analysis. We also demonstrate that the newly developed multicolor FISH method is compatible with an improved cell fixation protocol for FISH targeting Gram-negative bacterial populations. This fixation approach uses agarose embedding during formaldehyde fixation to better preserve the three-dimensional structure of spatially complex samples such as biofilms and activated sludge flocs. The new multicolor FISH approach should be highly suitable for studying structural and functional aspects of microbial communities in virtually all types of samples that can be analyzed by conventional FISH methods.
Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella basta.
2019 - Environ. Microbiol., 10: 3831-3854
Abstract:
Marine sponges represent one of the few eukaryotic groups that frequently harbour symbiotic members of the Thaumarchaeota, which are important chemoautotrophic ammonia-oxidizers in many environments. However, in most studies, direct demonstration of ammonia-oxidation by these archaea within sponges is lacking, and little is known about sponge-specific adaptations of ammonia-oxidizing archaea (AOA). Here, we characterized the thaumarchaeal symbiont of the marine sponge Ianthella basta using metaproteogenomics, fluorescence in situ hybridization, qPCR and isotope-based functional assays. 'Candidatus Nitrosospongia ianthellae' is only distantly related to cultured AOA. It is an abundant symbiont that is solely responsible for nitrite formation from ammonia in I. basta that surprisingly does not harbour nitrite-oxidizing microbes. Furthermore, this AOA is equipped with an expanded set of extracellular subtilisin-like proteases, a metalloprotease unique among archaea, as well as a putative branched-chain amino acid ABC transporter. This repertoire is strongly indicative of a mixotrophic lifestyle and is (with slight variations) also found in other sponge-associated, but not in free-living AOA. We predict that this feature as well as an expanded and unique set of secreted serpins (protease inhibitors), a unique array of eukaryotic-like proteins, and a DNA-phosporothioation system, represent important adaptations of AOA to life within these ancient filter-feeding animals.
Indications for enzymatic denitrification to N2O at low pH in an ammonia-oxidizing archaeon.
2019 - ISME J, 13: 2633-2638
Abstract:
Nitrous oxide (NO) is a key climate change gas and nitrifying microbes living in terrestrial ecosystems contribute significantly to its formation. Many soils are acidic and global change will cause acidification of aquatic and terrestrial ecosystems, but the effect of decreasing pH on NO formation by nitrifiers is poorly understood. Here, we used isotope-ratio mass spectrometry to investigate the effect of acidification on production of NO by pure cultures of two ammonia-oxidizing archaea (AOA; Nitrosocosmicus oleophilus and Nitrosotenuis chungbukensis) and an ammonia-oxidizing bacterium (AOB; Nitrosomonas europaea). For all three strains acidification led to increased emission of NO. However, changes of N site preference (SP) values within the NO molecule (as indicators of pathways for NO formation), caused by decreasing pH, were highly different between the tested AOA and AOB. While acidification decreased the SP value in the AOB strain, SP values increased to a maximum value of 29‰ in N. oleophilus. In addition, N-nitrite tracer experiments showed that acidification boosted nitrite transformation into NO in all strains, but the incorporation rate was different for each ammonia oxidizer. Unexpectedly, for N. oleophilus more than 50% of the NO produced at pH 5.5 had both nitrogen atoms from nitrite and we demonstrated that under these conditions expression of a putative cytochrome P450 NO reductase is strongly upregulated. Collectively, our results indicate that N. oleophilus might be able to enzymatically denitrify nitrite to NO at low pH.
Mitigating Anticipated Effects of Systematic Errors Supports Sister-Group Relationship between Xenacoelomorpha and Ambulacraria.
2019 - Curr. Biol., 11: 1818-1826.e6 Hiroaki Nakano, 2017, BMC Evolutionary Biology 17 (1), DOI:10.1186/s12862-017-1080-2, CC BY-SA 4.0")
Abstract:
Xenoturbella and the acoelomorph worms (Xenacoelomorpha) are simple marine animals with controversial affinities. They have been placed as the sister group of all other bilaterian animals (Nephrozoa hypothesis), implying their simplicity is an ancient characteristic; alternatively, they have been linked to the complex Ambulacraria (echinoderms and hemichordates) in a clade called the Xenambulacraria, suggesting their simplicity evolved by reduction from a complex ancestor. The difficulty resolving this problem implies the phylogenetic signal supporting the correct solution is weak and affected by inadequate modeling, creating a misleading non-phylogenetic signal. The idea that the Nephrozoa hypothesis might be an artifact is prompted by the faster molecular evolutionary rate observed within the Acoelomorpha. Unequal rates of evolution are known to result in the systematic artifact of long branch attraction, which would be predicted to result in an attraction between long-branch acoelomorphs and the outgroup, pulling them toward the root. Other biases inadequately accommodated by the models used can also have strong effects, exacerbated in the context of short internal branches and long terminal branches. We have assembled a large and informative dataset to address this problem. Analyses designed to reduce or to emphasize misleading signals show the Nephrozoa hypothesis is supported under conditions expected to exacerbate errors, and the Xenambulacraria hypothesis is preferred in conditions designed to reduce errors. Our reanalyses of two other recently published datasets produce the same result. We conclude that the Xenacoelomorpha are simplified relatives of the Ambulacraria.
Global diversity and biogeography of bacterial communities in wastewater treatment plants.
2019 - Nat Microbiol, 7: 1183-1195
Abstract:
Microorganisms in wastewater treatment plants (WWTPs) are essential for water purification to protect public and environmental health. However, the diversity of microorganisms and the factors that control it are poorly understood. Using a systematic global-sampling effort, we analysed the 16S ribosomal RNA gene sequences from ~1,200 activated sludge samples taken from 269 WWTPs in 23 countries on 6 continents. Our analyses revealed that the global activated sludge bacterial communities contain ~1 billion bacterial phylotypes with a Poisson lognormal diversity distribution. Despite this high diversity, activated sludge has a small, global core bacterial community (n = 28 operational taxonomic units) that is strongly linked to activated sludge performance. Meta-analyses with global datasets associate the activated sludge microbiomes most closely to freshwater populations. In contrast to macroorganism diversity, activated sludge bacterial communities show no latitudinal gradient. Furthermore, their spatial turnover is scale-dependent and appears to be largely driven by stochastic processes (dispersal and drift), although deterministic factors (temperature and organic input) are also important. Our findings enhance our mechanistic understanding of the global diversity and biogeography of activated sludge bacterial communities within a theoretical ecology framework and have important implications for microbial ecology and wastewater treatment processes.
Symbiont-mediated defense against Legionella pneumophila in amoebae
2019 - mBio, 10: e00333-19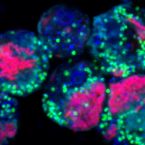
Abstract:
Legionella pneumophila is an important opportunistic pathogen for which environmental reservoirs are crucial for the infection of humans. In the environment, free-living amoebae represent key hosts providing nutrients and shelter for highly efficient intracellular proliferation of L. pneumophila, which eventually leads to lysis of the protist. However, the significance of other bacterial players for L. pneumophila ecology is poorly understood. In this study, we used a ubiquitous amoeba and bacterial endosymbiont to investigate the impact of this common association on L. pneumophilainfection. We demonstrate that L. pneumophila proliferation was severely suppressed in Acanthamoeba castellanii harboring the chlamydial symbiont Protochlamydia amoebophila. The amoebae survived the infection and were able to resume growth. Different environmental amoeba isolates containing the symbiont were equally well protected as different L. pneumophila isolates were diminished, suggesting ecological relevance of this symbiont-mediated defense. Furthermore, protection was not mediated by impaired L. pneumophila uptake. Instead, we observed reduced virulence of L. pneumophila released from symbiont-containing amoebae. Pronounced gene expression changes in the presence of the symbiont indicate that interference with the transition to the transmissive phase impedes the L. pneumophila infection. Finally, our data show that the defensive response of amoebae harboring P. amoebophila leaves the amoebae with superior fitness reminiscent of immunological memory. Given that mutualistic associations between bacteria and amoebae are widely distributed, P. amoebophila and potentially other amoeba endosymbionts could be key in shaping environmental survival, abundance, and virulence of this important pathogen, thereby affecting the frequency of human infection.
Cometabolic biotransformation and microbial-mediated abiotic transformation of sulfonamides by three ammonia oxidizers.
2019 - Water Res., 444-453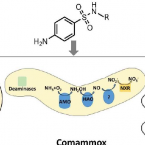
Abstract:
The abilities of three phylogenetically distant ammonia oxidizers, Nitrososphaera gargensis, an ammonia-oxidizing archaeon (AOA); Nitrosomomas nitrosa Nm90, an ammonia-oxidizing bacterium (AOB); and Nitrospira inopinata, the only complete ammonia oxidizer (comammox) available as a pure culture, to biotransform seven sulfonamides (SAs) were investigated. The removals and protein-normalized biotransformation rate constants indicated that the AOA strain N. gargensis exhibited the highest SA biotransformation rates, followed by N. inopinata and N. nitrosa Nm90. The transformation products (TPs) of sulfadiazine (SDZ), sulfamethazine (SMZ) and sulfamethoxazole (SMX) and the biotransformation mechanisms were evaluated. Based on the analysis of the TP formulas and approximate structures, it was found that during biotransformation, i) the AOA strain carried out SA deamination, hydroxylation, and nitration; ii) the AOB strain mainly performed SA deamination; and iii) the comammox isolate participated only in deamination reactions. It is proposed that deamination was catalyzed by deaminases while hydroxylation and nitration were mediated by nonspecific activities of the ammonia monooxygenase (AMO). Additionally, it was demonstrated that among the three ammonia oxidizers, only AOB contributed to the formation of pterin-SA conjugates. The biotransformation of SDZ, SMZ and SMX occurred only when ammonia oxidation was active, suggesting a cometabolic transformation mechanism. Interestingly, SAs could also be transformed by hydroxylamine, an intermediate of ammonia oxidation, suggesting that in addition to enzymatic conversions, a microbially induced abiotic mechanism contributes to SA transformation during ammonia oxidation. Overall, using experiments with pure cultures, this study provides important insights into the roles played by ammonia oxidizers in SA biotransformation.
Mucispirillum schaedleri antagonizes Salmonella virulence to protect mice against colitis
2019 - Cell Host Microbe, 25: 681-694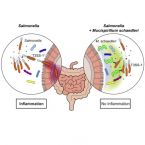
Abstract:
The microbiota and the gastrointestinal mucus layer play a pivotal role in protection against non-typhoidal Salmonellaenterica serovar Typhimurium (S. Tm) colitis. Here, we analyzed the course of Salmonella colitis in mice lacking a functional mucus layer in the gut. Unexpectedly, in contrast to mucus-proficient littermates, genetically deficient mice were protected against Salmonella-induced gut inflammation in the streptomycin colitis model. This correlated with microbiota alterations and enrichment of the bacterial phylum Deferribacteres. Using gnotobiotic mice associated with defined bacterial consortia, we causally linked Mucispirillum schaedleri, currently the sole known representative of Deferribacteres present in the mammalian microbiota, to host protection against S. Tm colitis. Inhibition by M. schaedleri involves interference with S. Tm invasion gene expression, partly by competing for anaerobic electron acceptors. In conclusion, this study establishes M. schaedleri, a core member of the murine gut microbiota, as a key antagonist of S. Tm virulence in the gut.
The cooling tower water microbiota: Seasonal dynamics and co-occurrence of bacterial and protist phylotypes.
2019 - Water Res., 464-479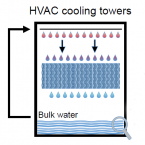
Abstract:
Cooling towers for heating, ventilation and air conditioning are ubiquitous in the built environment. Often located on rooftops, their semi-open water basins provide a suitable environment for microbial growth. They are recognized as a potential source of bacterial pathogens and have been associated with disease outbreaks such as Legionnaires' disease. While measures to minimize public health risks are in place, the general microbial and protist community structure and dynamics in these systems remain largely elusive. In this study, we analysed the microbiome of the bulk water from the basins of three cooling towers by 16S and 18S rRNA gene amplicon sequencing over the course of one year. Bacterial diversity in all three towers was broadly comparable to other freshwater systems, yet less diverse than natural environments; the most abundant taxa are also frequently found in freshwater or drinking water. While each cooling tower had a pronounced site-specific microbial community, taxa shared among all locations mainly included groups generally associated with biofilm formation. We also detected several groups related to known opportunistic pathogens, such as Legionella, Mycobacterium, and Pseudomonas species, albeit at generally low abundance. Although cooling towers represent a rather stable environment, microbial community composition was highly dynamic and subject to seasonal change. Protists are important members of the cooling tower water microbiome and known reservoirs for bacterial pathogens. Co-occurrence analysis of bacteria and protist taxa successfully captured known interactions between amoeba-associated bacteria and their hosts, and predicted a large number of additional relationships involving ciliates and other protists. Together, this study provides an unbiased and comprehensive overview of microbial diversity of cooling tower water basins, establishing a framework for investigating and assessing public health risks associated with these man-made freshwater environments.
Low yield and abiotic origin of N2O formed by the complete nitrifier Nitrospira inopinata.
2019 - Nat Commun, 1: 1836
Abstract:
Nitrous oxide (NO) and nitric oxide (NO) are atmospheric trace gases that contribute to climate change and affect stratospheric and ground-level ozone concentrations. Ammonia oxidizing bacteria (AOB) and archaea (AOA) are key players in the nitrogen cycle and major producers of NO and NO globally. However, nothing is known about NO and NO production by the recently discovered and widely distributed complete ammonia oxidizers (comammox). Here, we show that the comammox bacterium Nitrospira inopinata is sensitive to inhibition by an NO scavenger, cannot denitrify to NO, and emits NO at levels that are comparable to AOA but much lower than AOB. Furthermore, we demonstrate that NO formed by N. inopinata formed under varying oxygen regimes originates from abiotic conversion of hydroxylamine. Our findings indicate that comammox microbes may produce less NO during nitrification than AOB.
Widespread soil bacterium that oxidizes atmospheric methane.
2019 - Proc. Natl. Acad. Sci. U.S.A., 17: 8515-8524
Abstract:
The global atmospheric level of methane (CH), the second most important greenhouse gas, is currently increasing by ∼10 million tons per year. Microbial oxidation in unsaturated soils is the only known biological process that removes CH from the atmosphere, but so far, bacteria that can grow on atmospheric CH have eluded all cultivation efforts. In this study, we have isolated a pure culture of a bacterium, strain MG08 that grows on air at atmospheric concentrations of CH [1.86 parts per million volume (p.p.m.v.)]. This organism, named , is globally distributed in soils and closely related to uncultured members of the upland soil cluster α. CH oxidation experiments and C-single cell isotope analyses demonstrated that it oxidizes atmospheric CH aerobically and assimilates carbon from both CH and CO Its estimated specific affinity for CH (a) is the highest for any cultivated methanotroph. However, growth on ambient air was also confirmed for and , close relatives with a lower specific affinity for CH, suggesting that the ability to utilize atmospheric CH for growth is more widespread than previously believed. The closed genome of MG08 encodes a single particulate methane monooxygenase, the serine cycle for assimilation of carbon from CH and CO, and CO fixation via the recently postulated reductive glycine pathway. It also fixes dinitrogen and expresses the genes for a high-affinity hydrogenase and carbon monoxide dehydrogenase, suggesting that atmospheric CH oxidizers harvest additional energy from oxidation of the atmospheric trace gases carbon monoxide (0.2 p.p.m.v.) and hydrogen (0.5 p.p.m.v.).
An automated Raman-based platform for the sorting of live cells by functional properties.
2019 - Nat Microbiol, 6: 1035-1048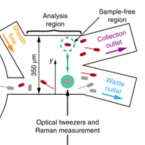
Abstract:
Stable-isotope probing is widely used to study the function of microbial taxa in their natural environment, but sorting of isotopically labelled microbial cells from complex samples for subsequent genomic analysis or cultivation is still in its early infancy. Here, we introduce an optofluidic platform for automated sorting of stable-isotope-probing-labelled microbial cells, combining microfluidics, optical tweezing and Raman microspectroscopy, which yields live cells suitable for subsequent single-cell genomics, mini-metagenomics or cultivation. We describe the design and optimization of this Raman-activated cell-sorting approach, illustrate its operation with four model bacteria (two intestinal, one soil and one marine) and demonstrate its high sorting accuracy (98.3 ± 1.7%), throughput (200-500 cells h; 3.3-8.3 cells min) and compatibility with cultivation. Application of this sorting approach for the metagenomic characterization of bacteria involved in mucin degradation in the mouse colon revealed a diverse consortium of bacteria, including several members of the underexplored family Muribaculaceae, highlighting both the complexity of this niche and the potential of Raman-activated cell sorting for identifying key players in targeted processes.
Resolving the individual contribution of key microbial populations to enhanced biological phosphorus removal with Raman-FISH.
2019 - ISME J, 8: 1933-1946
Abstract:
Enhanced biological phosphorus removal (EBPR) is a globally important biotechnological process and relies on the massive accumulation of phosphate within special microorganisms. Candidatus Accumulibacter conform to the classical physiology model for polyphosphate accumulating organisms and are widely believed to be the most important player for the process in full-scale EBPR systems. However, it was impossible till now to quantify the contribution of specific microbial clades to EBPR. In this study, we have developed a new tool to directly link the identity of microbial cells to the absolute quantification of intracellular poly-P and other polymers under in situ conditions, and applied it to eight full-scale EBPR plants. Besides Ca. Accumulibacter, members of the genus Tetrasphaera were found to be important microbes for P accumulation, and in six plants they were the most important. As these Tetrasphaera cells did not exhibit the classical phenotype of poly-P accumulating microbes, our entire understanding of the microbiology of the EBPR process has to be revised. Furthermore, our new single-cell approach can now also be applied to quantify storage polymer dynamics in individual populations in situ in other ecosystems and might become a valuable tool for many environmental microbiologists.
Historical factors associated with past environments influence the biogeography of thermophilic endospores in Arctic marine sediments
2019 - Front Microbiol, 10: 245
Abstract:
Selection by the local, contemporary environment plays a prominent role in shaping the biogeography of microbes. However, the importance of historical factors in microbial biogeography is more debatable. Historical factors include past ecological and evolutionary circumstances that may have influenced present-day microbial diversity, such as dispersal and past environmental conditions. Diverse thermophilic sulfate-reducing are present as dormant endospores in marine sediments worldwide where temperatures are too low to support their growth. Therefore, they are dispersed to here from elsewhere, presumably a hot, anoxic habitat. While dispersal through ocean currents must influence their distribution in cold marine sediments, it is not clear whether even earlier historical factors, related to the source habitat where these organisms were once active, also have an effect. We investigated whether these historical factors may have influenced the diversity and distribution of thermophilic endospores by comparing their diversity in 10 Arctic fjord surface sediments. Although community composition varied spatially, clear biogeographic patterns were only evident at a high level of taxonomic resolution (>97% sequence similarity of the 16S rRNA gene) achieved with oligotyping. In particular, the diversity and distribution of oligotypes differed for the two most prominent OTUs (defined using a standard 97% similarity cutoff). One OTU was dominated by a single ubiquitous oligotype, while the other OTU consisted of ten more spatially localized oligotypes that decreased in compositional similarity with geographic distance. These patterns are consistent with differences in historical factors that occurred when and where the taxa were once active, prior to sporulation. Further, the influence of history on biogeographic patterns was only revealed by analyzing microdiversity within OTUs, suggesting that populations within standard OTU-level groupings do not necessarily share a common ecological and evolutionary history.
Dark aerobic sulfide oxidation by anoxygenic phototrophs in anoxic waters.
2019 - Environ. Microbiol., 5: 1611-1626
Abstract:
Anoxygenic phototrophic sulfide oxidation by green and purple sulfur bacteria (PSB) plays a key role in sulfide removal from anoxic shallow sediments and stratified waters. Although some PSB can also oxidize sulfide with nitrate and oxygen, little is known about the prevalence of this chemolithotrophic lifestyle in the environment. In this study, we investigated the role of these phototrophs in light-independent sulfide removal in the chemocline of Lake Cadagno. Our temporally resolved, high-resolution chemical profiles indicated that dark sulfide oxidation was coupled to high oxygen consumption rates of ~9 μM O ·h . Single-cell analyses of lake water incubated with CO in the dark revealed that Chromatium okenii was to a large extent responsible for aerobic sulfide oxidation and it accounted for up to 40% of total dark carbon fixation. The genome of Chr. okenii reconstructed from the Lake Cadagno metagenome confirms its capacity for microaerophilic growth and provides further insights into its metabolic capabilities. Moreover, our genomic and single-cell data indicated that other PSB grow microaerobically in these apparently anoxic waters. Altogether, our observations suggest that aerobic respiration may not only play an underappreciated role in anoxic environments but also that organisms typically considered strict anaerobes may be involved.
Rapid transfer of plant photosynthates to soil bacteria via ectomycorrhizal hyphae and its interaction with nitrogen availability.
2019 - Front Microbiol, 168
Abstract:
Plant roots release recent photosynthates into the rhizosphere, accelerating decomposition of organic matter by saprotrophic soil microbes ("rhizosphere priming effect") which consequently increases nutrient availability for plants. However, about 90% of all higher plant species are mycorrhizal, transferring a significant fraction of their photosynthates directly to their fungal partners. Whether mycorrhizal fungi pass on plant-derived carbon (C) to bacteria in root-distant soil areas, i.e., incite a "hyphosphere priming effect," is not known. Experimental evidence for C transfer from mycorrhizal hyphae to soil bacteria is limited, especially for ectomycorrhizal systems. As ectomycorrhizal fungi possess enzymatic capabilities to degrade organic matter themselves, it remains unclear whether they cooperate with soil bacteria by providing photosynthates, or compete for available nutrients. To investigate a possible C transfer from ectomycorrhizal hyphae to soil bacteria, and its response to changing nutrient availability, we planted young beech trees () into "split-root" boxes, dividing their root systems into two disconnected soil compartments. Each of these compartments was separated from a litter compartment by a mesh penetrable for fungal hyphae, but not for roots. Plants were exposed to a C-CO-labeled atmosphere, while N-labeled ammonium and amino acids were added to one side of the split-root system. We found a rapid transfer of recent photosynthates via ectomycorrhizal hyphae to bacteria in root-distant soil areas. Fungal and bacterial phospholipid fatty acid (PLFA) biomarkers were significantly enriched in hyphae-exclusive compartments 24 h after C-CO-labeling. Isotope imaging with nanometer-scale secondary ion mass spectrometry (NanoSIMS) allowed for the first time visualization of plant-derived C and N taken up by an extraradical fungal hypha, and in microbial cells thriving on hyphal surfaces. When N was added to the litter compartments, bacterial biomass, and the amount of incorporated C strongly declined. Interestingly, this effect was also observed in adjacent soil compartments where added N was only available for bacteria through hyphal transport, indicating that ectomycorrhizal fungi were acting on soil bacteria. Together, our results demonstrate that (i) ectomycorrhizal hyphae rapidly transfer plant-derived C to bacterial communities in root-distant areas, and (ii) this transfer promptly responds to changing soil nutrient conditions.
Long-term transcriptional activity at zero growth by a cosmopolitan rare biosphere member
2019 - mBio, 10: e02189-18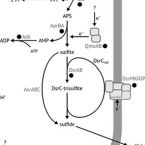
Abstract:
Microbial diversity in the environment is mainly concealed within the rare biosphere (all species with <0.1% relative abundance). While dormancy explains a low-abundance state very well, the mechanisms leading to rare but active microorganisms remain elusive. We used environmental systems biology to genomically and transcriptionally characterise Candidatus Desulfosporosinus infrequens, a low-abundance sulfate-reducing microorganism cosmopolitan to freshwater wetlands, where it contributes to cryptic sulfur cycling. We obtained its near-complete genome by metagenomics of acidic peat soil. In addition, we analyzed anoxic peat soil incubated under in situ-like conditions for 50 days by Desulfosporosinus-targeted qPCR and metatranscriptomics. The Desulfosporosinus population stayed at a constant low abundance under all incubation conditions, averaging 1.2 × 10⁶ 16S rRNA gene copies per cm³ soil. In contrast, transcriptional activity of Ca.D. infrequens increased at day 36 by 56- to 188-fold when minor amendments of acetate, propionate, lactate, or butyrate were provided with sulfate, as compared to the no-substrate-control. Overall transcriptional activity was driven by expression of genes encoding ribosomal proteins, energy metabolism and stress response but not by expression of genes encoding cell growth-associated processes. Since our results did not support growth of these highly active microorganisms in terms of biomass increase or cell division, they had to invest their sole energy for maintenance, most likely counterbalancing acidic pH conditions. This finding explains how a rare biosphere member can contribute to a biogeochemically relevant process while remaining in a zero growth state over a period of 50 days.
Surface-enhanced Raman spectroscopy of microorganisms: limitations and applicability on the single-cell level.
2019 - Analyst, 3: 943-953
Abstract:
Detection and characterization of microorganisms is essential for both clinical diagnostics and environmental studies. An emerging technique to analyse microbes at single-cell resolution is surface-enhanced Raman spectroscopy (surface-enhanced Raman scattering: SERS). Optimised SERS procedures enable fast analytical read-outs with specific molecular information, providing insight into the chemical composition of microbiological samples. Knowledge about the origin of microbial SERS signals and parameter(s) affecting their occurrence, intensity and/or reproducibility is crucial for reliable SERS-based analyses. In this work, we explore the feasibility and limitations of the SERS approach for characterizing microbial cells and investigate the applicability of SERS for single-cell sorting as well as for three-dimensional visualization of microbial communities. Analyses of six different microbial species (an archaeon, two Gram-positive bacteria, three Gram-negative bacteria) showed that for several of these organisms distinct features in their SERS spectra were lacking. As additional confounding factor, the physiological conditions of the cells (as influenced by e.g., storage conditions or deuterium-labelling) were systematically addressed, for which we conclude that the respective SERS signal at the single-cell level is strongly influenced by the metabolic activity of the analysed cells. While this finding complicates the interpretation of SERS data, it may on the other hand enable probing of the metabolic state of individual cells within microbial populations of interest.
Cyanate and urea are substrates for nitrification by Thaumarchaeota in the marine environment.
2019 - Nat Microbiol, 2: 234-243
Abstract:
Ammonia-oxidizing archaea of the phylum Thaumarchaeota are among the most abundant marine microorganisms. These organisms thrive in the oceans despite ammonium being present at low nanomolar concentrations. Some Thaumarchaeota isolates have been shown to utilize urea and cyanate as energy and N sources through intracellular conversion to ammonium. Yet, it is unclear whether patterns observed in culture extend to marine Thaumarchaeota, and whether Thaumarchaeota in the ocean directly utilize urea and cyanate or rely on co-occurring microorganisms to break these substrates down to ammonium. Urea utilization has been reported for marine ammonia-oxidizing communities, but no evidence of cyanate utilization exists for marine ammonia oxidizers. Here, we demonstrate that in the Gulf of Mexico, Thaumarchaeota use urea and cyanate both directly and indirectly as energy and N sources. We observed substantial and linear rates of nitrite production from urea and cyanate additions, which often persisted even when ammonium was added to micromolar concentrations. Furthermore, single-cell analysis revealed that the Thaumarchaeota incorporated ammonium-, urea- and cyanate-derived N at significantly higher rates than most other microorganisms. Yet, no cyanases were detected in thaumarchaeal genomic data from the Gulf of Mexico. Therefore, we tested cyanate utilization in Nitrosopumilus maritimus, which also lacks a canonical cyanase, and showed that cyanate was oxidized to nitrite. Our findings demonstrate that marine Thaumarchaeota can use urea and cyanate as both an energy and N source. On the basis of these results, we hypothesize that urea and cyanate are substrates for ammonia-oxidizing Thaumarchaeota throughout the ocean.
Sulfate is transported at significant rates through the symbiosome membrane and is crucial for nitrogenase biosynthesis.
2019 - Plant Cell Environ., 4: 1180-1189
Abstract:
Legume-rhizobia symbioses play a major role in food production for an ever growing human population. In this symbiosis, dinitrogen is reduced ("fixed") to ammonia by the rhizobial nitrogenase enzyme complex and is secreted to the plant host cells, whereas dicarboxylic acids derived from photosynthetically produced sucrose are transported into the symbiosomes and serve as respiratory substrates for the bacteroids. The symbiosome membrane contains high levels of SST1 protein, a sulfate transporter. Sulfate is an essential nutrient for all living organisms, but its importance for symbiotic nitrogen fixation and nodule metabolism has long been underestimated. Using chemical imaging, we demonstrate that the bacteroids take up 20-fold more sulfate than the nodule host cells. Furthermore, we show that nitrogenase biosynthesis relies on high levels of imported sulfate, making sulfur as essential as carbon for the regulation and functioning of symbiotic nitrogen fixation. Our findings thus establish the importance of sulfate and its active transport for the plant-microbe interaction that is most relevant for agriculture and soil fertility.
Transcriptomic and proteomic insight into the mechanism of cyclooctasulfur- versus thiosulfate-oxidation by the chemolithoautotroph Sulfurimonas denitrificans.
2019 - Environ. Microbiol., 1: 244-258
Abstract:
Chemoautotrophic bacteria belonging to the genus Sulfurimonas (class Campylobacteria) were previously identified as key players in the turnover of zero-valence sulfur, a central intermediate in the marine sulfur cycle. S. denitrificans was further shown to be able to oxidize cyclooctasulfur (S ). However, at present the mechanism of activation and metabolism of cyclooctasulfur is not known. Here, we assessed the transcriptome and proteome of S. denitrificans grown with either thiosulfate or S as the electron donor. While the overall expression profiles under the two growth conditions were rather similar, distinct differences were observed that could be attributed to the utilization of S . This included a higher abundance of expressed genes related to surface attachment in the presence of S , and the differential regulation of the sulfur-oxidation multienzyme complex (SOX), which in S. denitrificans is encoded in two gene clusters: soxABXY Z and soxCDY Z . While the proteins of both clusters were present with thiosulfate, only proteins of the soxCDY Z were detected at significant levels with S . Based on these findings a model for the oxidation of S is proposed. Our results have implications for interpreting metatranscriptomic and -proteomic data and for the observed high level of diversification of soxY Z among sulfur-oxidizing Campylobacteria.
Book chapters and other publications
Population Genomics: Microorganisms
2019 - in Population Genomics: Microorganisms. (Polz MF, Rajora O). Nature Springer
Mikrobiome – Wissensstand und Perspektiven
2019 - 17-29. in Die unbekannte Welt der Mikrobiome; Rundgespräche Forum Ökologie Bd. 47. (Bauer J & von Mutius E). Bayerische Akademie der Wissenschaften; Verlag Dr. Friedrich Pfeil


Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at