Publications
Publications in peer reviewed journals
Sedimentary sulfides
2017 - Elements, 13: 117–122.
Abstract:
Sedimentary sulfides constitute over 95% of the sulfide on the surface
of the planet, and their formation, preservation and destruction largely
determines the surface environment. The sulfide in sediments is mainly
derived from the products of sulfate-reducing bacteria, which are currently
responsible for oxidizing over half the organic matter flux reaching sediments.
Pyrite is the mineral overwhelmingly produced. The geochemistry of pyrite,
both in terms of its isotopic composition and its trace-element loading, has
varied dramatically over geologic time. As such, it is a major source of our
current understanding about the nature of the early Earth and of the Earth’s
subsequent geochemical and biological evolution.The life sulfuric: microbial ecology of sulfur cycling in marine sediments.
2017 - Environ Microbiol Rep, 4: 323-344
Abstract:
Almost the entire seafloor is covered with sediments that can be more than 10 000 m thick and represent a vast microbial ecosystem that is a major component of Earth's element and energy cycles. Notably, a significant proportion of microbial life in marine sediments can exploit energy conserved during transformations of sulfur compounds among different redox states. Sulfur cycling, which is primarily driven by sulfate reduction, is tightly interwoven with other important element cycles (carbon, nitrogen, iron, manganese) and therefore has profound implications for both cellular- and ecosystem-level processes. Sulfur-transforming microorganisms have evolved diverse genetic, metabolic, and in some cases, peculiar phenotypic features to fill an array of ecological niches in marine sediments. Here, we review recent and selected findings on the microbial guilds that are involved in the transformation of different sulfur compounds in marine sediments and emphasise how these are interlinked and have a major influence on ecology and biogeochemistry in the seafloor. Extraordinary discoveries have increased our knowledge on microbial sulfur cycling, mainly in sulfate-rich surface sediments, yet many questions remain regarding how sulfur redox processes may sustain the deep-subsurface biosphere and the impact of organic sulfur compounds on the marine sulfur cycle.
Direct single-cell biomass estimates for marine bacteria via Archimedes' principle.
2017 - ISME J, 3: 825-828
Abstract:
Microbes are an essential component of marine food webs and biogeochemical cycles, and therefore precise estimates of their biomass are of significant value. Here, we measured single-cell biomass distributions of isolates from several numerically abundant marine bacterial groups, including Pelagibacter (SAR11), Prochlorococcus and Vibrio using a microfluidic mass sensor known as a suspended microchannel resonator (SMR). We show that the SMR can provide biomass (dry mass) measurements for cells spanning more than two orders of magnitude and that these estimates are consistent with other independent measures. We find that Pelagibacterales strain HTCC1062 has a median biomass of 11.9±0.7 fg per cell, which is five- to twelve-fold smaller than the median Prochlorococcus cell's biomass (depending upon strain) and nearly 100-fold lower than that of rapidly growing V. splendidus strain 13B01. Knowing the biomass contributions from various taxonomic groups will provide more precise estimates of total marine biomass, aiding models of nutrient flux in the ocean.
Vibrio crassostreae, a benign oyster colonizer turned into a pathogen after plasmid acquisition.
2017 - ISME J, 4: 1043-1052
Abstract:
Vibrios are frequently associated with oyster mortality; however whether they are the primary causative agent or secondary opportunistic colonizers is not well understood. Here we combine analysis of natural infection dynamics, population genomics and molecular genetics to ask (i) to what extent oysters are passively colonized by Vibrio population present in the surrounding water, (ii) how populations turn over during pathogenicity events and (iii) what genetic factors are responsible for pathogenicity. We identified several populations of Vibrio preferentially associated with oyster tissues. Among these, Vibrio crassostreae is particularly abundant in diseased animals while nearly absent in the surrounding water, and its pathogenicity is correlated with the presence of a large mobilizable plasmid. We further demonstrate that the plasmid is essential for killing but not necessary for survival in tissues of oysters. Our results suggest that V. crassostreae first differentiated into a benign oyster colonizer that was secondarily turned into a pathogen by introgression of a virulence plasmid into the population, possibly facilitated by elevated host density in farming areas.
Physiological and gene expression responses to nitrogen regimes and temperatures in Mastigocladus sp. strain CHP1, a predominant thermotolerant cyanobacterium of hot springs.
2017 - Syst. Appl. Microbiol., 2: 102-113
Abstract:
Cyanobacteria are widely distributed primary producers with significant implications for the global biogeochemical cycles of carbon and nitrogen. Diazotrophic cyanobacteria of subsection V (Order Stigonematales) are particularly ubiquitous in photoautotrophic microbial mats of hot springs. The Stigonematal cyanobacterium strain CHP1 isolated from the Porcelana hot spring (Chile) was one of the major contributors of the new nitrogen through nitrogen fixation. Further morphological and genetic characterization verified that the strain CHP1 belongs to Stigonematales, and it formed a separate clade together with other thermophiles of the genera Fischerella and Mastigocladus. Strain CHP1 fixed maximum N in the light, independent of the temperature range. At 50°C nifH gene transcripts showed high expression during the light period, whereas the nifH gene expression at 45°C was arrhythmic. The strain displayed a high affinity for nitrate and a low tolerance for high ammonium concentrations, whereas the narB and glnA genes showed higher expression in light and at the beginning of the dark phase. It is proposed that Mastigocladus sp. strain CHP1 would represent a good model for the study of subsection V thermophilic cyanobacteria, and for understanding the adaptations of these photoautotrophic organisms inhabiting microbial mats in hot springs globally.
Endemicity of the cosmopolitan mesophilic chemolithoautotroph Sulfurimonas at deep-sea hydrothermal vents.
2017 - ISME J, 4: 909-919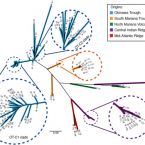
Abstract:
Rich animal and microbial communities have been found at deep-sea hydrothermal vents. Although the biogeography of vent macrofauna is well understood, the corresponding knowledge about vent microbial biogeography is lacking. Here, we apply the multilocus sequence analysis (MLSA) to assess the genetic variation of 109 Sulfurimonas strains with ⩾98% 16S rRNA gene sequence similarity, which were isolated from four different geographical regions (Okinawa Trough (OT), Mariana Volcanic Arc and Trough (MVAT), Central Indian Ridge (CIR) and Mid-Atlantic Ridge (MAR)). Sequence typing based on 11 protein-coding genes revealed high genetic variation, including some allele types that are widespread within regions, resulting in 102 nucleotide sequence types (STs). This genetic variation was predominantly due to mutation rather than recombination. Phylogenetic analysis of the 11 concatenated genes showed a clear geographical isolation corresponding to the hydrothermal regions they originated from, suggesting limited dispersal. Genetic differentiation among Sulfurimonas populations was primarily influenced by geographical distance rather than gas composition of vent fluid or habitat, although in situ environmental conditions of each microhabitat could not be examined. Nevertheless, Sulfurimonas may possess a higher dispersal capability compared with deep-sea hydrothermal vent thermophiles. This is the first report on MLSA of deep-sea hydrothermal vent Epsilonproteobacteria, which is indicative of allopatric speciation.
Microbial community assembly and evolution in subseafloor sediment.
2017 - Proc. Natl. Acad. Sci. U.S.A., 11: 2940-2945
Abstract:
Bacterial and archaeal communities inhabiting the subsurface seabed live under strong energy limitation and have growth rates that are orders of magnitude slower than laboratory-grown cultures. It is not understood how subsurface microbial communities are assembled and whether populations undergo adaptive evolution or accumulate mutations as a result of impaired DNA repair under such energy-limited conditions. Here we use amplicon sequencing to explore changes of microbial communities during burial and isolation from the surface to the >5,000-y-old subsurface of marine sediment and identify a small core set of mostly uncultured bacteria and archaea that is present throughout the sediment column. These persisting populations constitute a small fraction of the entire community at the surface but become predominant in the subsurface. We followed patterns of genome diversity with depth in four dominant lineages of the persisting populations by mapping metagenomic sequence reads onto single-cell genomes. Nucleotide sequence diversity was uniformly low and did not change with age and depth of the sediment. Likewise, there was no detectable change in mutation rates and efficacy of selection. Our results indicate that subsurface microbial communities predominantly assemble by selective survival of taxa able to persist under extreme energy limitation.
A Mobile Element in mutS Drives Hypermutation in a Marine Vibrio.
2017 - mBio, 1: in press
Abstract:
Bacteria face a trade-off between genetic fidelity, which reduces deleterious mistakes in the genome, and genetic innovation, which allows organisms to adapt. Evidence suggests that many bacteria balance this trade-off by modulating their mutation rates, but few mechanisms have been described for such modulation. Following experimental evolution and whole-genome resequencing of the marine bacterium Vibrio splendidus 12B01, we discovered one such mechanism, which allows this bacterium to switch to an elevated mutation rate. This switch is driven by the excision of a mobile element residing in mutS, which encodes a DNA mismatch repair protein. When integrated within the bacterial genome, the mobile element provides independent promoter and translation start sequences for mutS-different from the bacterium's original mutS promoter region-which allow the bacterium to make a functional mutS gene product. Excision of this mobile element rejoins the mutS gene with host promoter and translation start sequences but leaves a 2-bp deletion in the mutS sequence, resulting in a frameshift and a hypermutator phenotype. We further identified hundreds of clinical and environmental bacteria across Betaproteobacteria and Gammaproteobacteria that possess putative mobile elements within the same amino acid motif in mutS In a subset of these bacteria, we detected excision of the element but not a frameshift mutation; the mobile elements leave an intact mutS coding sequence after excision. Our findings reveal a novel mechanism by which one bacterium alters its mutation rate and hint at a possible evolutionary role for mobile elements within mutS in other bacteria.
DNA mutations are a double-edged sword. Most mutations are harmful; they can scramble precise genetic sequences honed over thousands of generations. However, in rare cases, mutations also produce beneficial new traits that allow populations to adapt to changing environments. Recent evidence suggests that some bacteria balance this trade-off by altering their mutation rates to suit their environment. To date, however, we know of few mechanisms that allow bacteria to change their mutation rates. We describe one such mechanism, driven by the action of a mobile element, in the marine bacterium Vibrio splendidus 12B01. We also found similar mobile genetic sequences in the mutS genes of many different bacteria, including clinical and agricultural pathogens. These mobile elements might play an as yet unknown role in the evolution of these important bacteria.Accurate Quantification of Laminarin in Marine Organic Matter with Enzymes from Marine Microbes.
2017 - Appl. Environ. Microbiol., 9: in press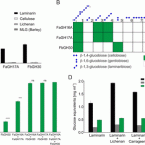
Abstract:
Marine algae produce a variety of glycans, which fulfill diverse biological functions and fuel the carbon and energy demands of heterotrophic microbes. A common approach to analysis of marine organic matter uses acid to hydrolyze the glycans into measurable monosaccharides. The monosaccharides may be derived from different glycans that are built with the same monosaccharides, however, and this approach does not distinguish between glycans in natural samples. Here we use enzymes to digest selectively and thereby quantify laminarin in particulate organic matter. Environmental metaproteome data revealed carbohydrate-active enzymes from marine flavobacteria as tools for selective hydrolysis of the algal β-glucan laminarin. The enzymes digested laminarin into glucose and oligosaccharides, which we measured with standard methods to establish the amounts of laminarin in the samples. We cloned, expressed, purified, and characterized three new glycoside hydrolases (GHs) of bacteria: two are endo-β-1,3-glucanases, of the GH16 and GH17 families, and the other is a GH30 exo-β-1,6-glucanase. sp. nov strain Hel1_33_131 GH30 (FbGH30) removed the β-1,6-glucose side chains, and GH17A (FaGH17A) and FaGH16A hydrolyzed the β-1,3-glucose backbone of laminarin. Specificity profiling with a library of glucan oligosaccharides and polysaccharides revealed that FaGH17A and FbGH30 were highly specific enzymes, while FaGH16A also hydrolyzed mixed-linked glucans with β-1,4-glucose. Therefore, we chose the more specific FaGH17A and FbGH30 to quantify laminarin in two cultured diatoms, namely, and , and in seawater samples from the North Sea and the Arctic Ocean. Combined, these results demonstrate the potential of enzymes for faster, stereospecific, and sequence-specific analysis of select glycans in marine organic matter. Marine algae synthesize substantial amounts of the glucose polymer laminarin for energy and carbon storage. Its concentrations, rates of production by autotrophic organisms, and rates of digestion by heterotrophic organisms remain unknown. Here we present a method based on enzymes that hydrolyze laminarin and enable its quantification even in crude substrate mixtures, without purification. Compared to the commonly used acid hydrolysis, the enzymatic method presented here is faster and stereospecific and selectively cleaves laminarin in mixtures of glycans, releasing only glucose and oligosaccharides, which can be easily quantified with reducing sugar assays.
Natural resource landscapes of a marine bacterium reveal distinct fitness-determining genes across the genome.
2017 - Environ. Microbiol., 6: 2422-2433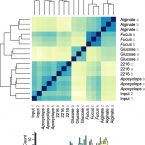
Abstract:
Heterotrophic bacteria exploit diverse microhabitats in the ocean, from particles to transient gradients. Yet the degree to which genes and pathways can contribute to an organism's fitness on such complex and variable natural resource landscapes remains poorly understood. Here, we determine the gene-by-gene fitness of a generalist saprophytic marine bacterium (Vibrio sp. F13 9CS106) on complex resources derived from its natural habitats - copepods (Apocyclops royi) and brown algae (Fucus vesiculosus) - and as reference substrates, glucose and the polysaccharide alginate, derived from brown algal cell walls. We find that resource complexity strongly buffers fitness costs of mutations, and that anabolic rather than catabolic pathways are more stringently required, likely due to functional redundancy in the latter. Moreover, while carbohydrate-rich algae requires several synthesis pathways, protein-rich Apocyclops does not, suggesting this ancestral habitat for Vibrios is a replete medium with metabolically redundant substrates. We also identify a candidate fitness trade-off for algal colonization: deletion of mshA increases mutant fitness. Our results demonstrate that gene fitness depends on habitat composition, and suggest that this generalist uses distinct resources in different natural habitats. The results further indicate that substrate replete conditions may lead to relatively relaxed selection on catabolic genes.
Exploiting fine-scale genetic and physiological variation of closely related microbes to reveal unknown enzyme functions.
2017 - J. Biol. Chem., 31: 13056-13067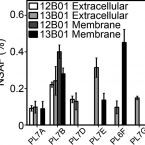
Abstract:
Polysaccharide degradation by marine microbes represents one of the largest and most rapid heterotrophic transformations of organic matter in the environment. Microbes employ systems of complementary carbohydrate-specific enzymes to deconstruct algal or plant polysaccharides (glycans) into monosaccharides. Because of the high diversity of glycan substrates, the functions of these enzymes are often difficult to establish. One solution to this problem may lie within naturally occurring microdiversity; varying numbers of enzymes, due to gene loss, duplication, or transfer, among closely related environmental microbes create metabolic differences akin to those generated by knock-out strains engineered in the laboratory used to establish the functions of unknown genes. Inspired by this natural fine-scale microbial diversity, we show here that it can be used to develop hypotheses guiding biochemical experiments for establishing the role of these enzymes in nature. In this work, we investigated alginate degradation among closely related strains of the marine bacterium One strain, 13B01, exhibited high extracellular alginate lyase activity compared with other strains. To identify the enzymes responsible for this high extracellular activity, we compared 13B01 with the previously characterized 12B01, which has low extracellular activity and lacks two alginate lyase genes present in 13B01. Using a combination of genomics, proteomics, biochemical, and functional screening, we identified a polysaccharide lyase family 7 enzyme that is unique to 13B01, secreted, and responsible for the rapid digestion of extracellular alginate. These results demonstrate the value of querying the enzymatic repertoires of closely related microbes to rapidly pinpoint key proteins with beneficial functions.
Adaptability as the key to success for the ubiquitous marine nitrite oxidizer Nitrococcus
2017 - Sci Adv, 3: e1700807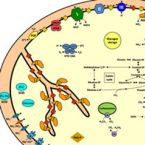
Abstract:
Nitrite-oxidizing bacteria (NOB) have conventionally been regarded as a highly specialized functional group responsible for the production of nitrate in the environment. However, recent culture-based studies suggest that they have the capacity to lead alternative lifestyles, but direct environmental evidence for the contribution of marine nitrite oxidizers to other processes has been lacking to date. We report on the alternative biogeochemical functions, worldwide distribution, and sometimes high abundance of the marine NOB Nitrococcus. These largely overlooked bacteria are capable of not only oxidizing nitrite but also reducing nitrate and producing nitrous oxide, an ozone-depleting agent and greenhouse gas. Furthermore, Nitrococcus can aerobically oxidize sulfide, thereby also engaging in the sulfur cycle. In the currently fast-changing global oceans, these findings highlight the potential functional switches these ubiquitous bacteria can perform in various biogeochemical cycles, each with distinct or even contrasting consequences.
Vitamin and Amino Acid Auxotrophy in Anaerobic Consortia Operating under Methanogenic Conditions.
2017 - mSystems, 5: e00038-17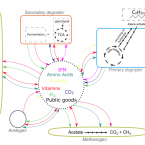
Abstract:
Syntrophy among Archaea and Bacteria facilitates the anaerobic degradation of organic compounds to CH4 and CO2. Particularly during aliphatic and aromatic hydrocarbon mineralization, as in the case of crude oil reservoirs and petroleum-contaminated sediments, metabolic interactions between obligate mutualistic microbial partners are of central importance. Using micromanipulation combined with shotgun metagenomic approaches, we describe the genomes of complex consortia within short-chain alkane-degrading cultures operating under methanogenic conditions. Metabolic reconstruction revealed that only a small fraction of genes in the metagenome-assembled genomes encode the capacity for fermentation of alkanes facilitated by energy conservation linked to H2 metabolism. Instead, the presence of inferred lifestyles based on scavenging anabolic products and intermediate fermentation products derived from detrital biomass was a common feature. Additionally, inferred auxotrophy for vitamins and amino acids suggests that the hydrocarbon-degrading microbial assemblages are structured and maintained by multiple interactions beyond the canonical H2-producing and syntrophic alkane degrader-methanogen partnership. Compared to previous work, our report points to a higher order of complexity in microbial consortia engaged in anaerobic hydrocarbon transformation. IMPORTANCE Microbial interactions between Archaea and Bacteria mediate many important chemical transformations in the biosphere from degrading abundant polymers to synthesis of toxic compounds. Two of the most pressing issues in microbial interactions are how consortia are established and how we can modulate these microbial communities to express desirable functions. Here, we propose that public goods (i.e., metabolites of high energy demand in biosynthesis) facilitate energy conservation for life under energy-limited conditions and determine the assembly and function of the consortia. Our report suggests that an understanding of public good dynamics could result in new ways to improve microbial pollutant degradation in anaerobic systems.
Allspice and Clove As Source of Triterpene Acids Activating the G Protein-Coupled Bile Acid Receptor TGR5.
2017 - Front Pharmacol, 8: 468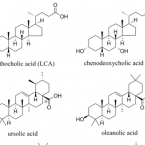
Abstract:
Worldwide, metabolic diseases such as obesity and type 2 diabetes have reached epidemic proportions. A major regulator of metabolic processes that gained interest in recent years is the bile acid receptor TGR5 (Takeda G protein-coupled receptor 5). This G protein-coupled membrane receptor can be found predominantly in the intestine, where it is mainly responsible for the secretion of the incretins glucagon-like peptide 1 (GLP-1) and peptide YY (PYY). The aim of this study was (i) to identify plant extracts with TGR5-activating potential, (ii) to narrow down their activity to the responsible constituents, and (iii) to assess whether the intestinal microbiota produces transformed metabolites with a different activity profile. Chenodeoxycholic acid (CDCA) served as positive control for both, the applied cell-based luciferase reporter gene assay for TGR5 activity and the biotransformation assay using mouse fecal slurry. The suitability of the workflow was demonstrated by the biotransformation of CDCA to lithocholic acid resulting in a distinct increase in TGR5 activity. Based on a traditional Tibetan formula, 19 plant extracts were selected and investigated for TGR5 activation. Extracts from the commonly used spices Syzygium aromaticum (SaroE, clove), Pimenta dioica (PdioE, allspice), and Kaempferia galanga (KgalE, aromatic ginger) significantly increased TGR5 activity. After biotransformation, only KgalE showed significant differences in its metabolite profile, which, however, did not alter its TGR5 activity compared to non-transformed KgalE. UHPLC-HRMS (high-resolution mass spectrometry) analysis revealed triterpene acids (TTAs) as the main constituents of the extracts SaroE and PdioE. Identification and quantification of TTAs in these two extracts as well as comparison of their TGR5 activity with reconstituted TTA mixtures allowed the attribution of the TGR5 activity to TTAs. EC50s were determined for the main TTAs, i.e., oleanolic acid (2.2 ± 1.6 μM), ursolic acid (1.1 ± 0.2 μM), as well as for the hitherto unknown TGR5 activators corosolic acid (0.5 ± 1.0 μM) and maslinic acid (3.7 ± 0.7 μM). In conclusion, extracts of clove, allspice, and aromatic ginger activate TGR5, which might play a pivotal role in their therapeutic use for the treatment of metabolic diseases. Moreover, the TGR5 activation of SaroE and PdioE could be pinpointed solely to TTAs.
Ammonia-oxidising archaea living at low pH: Insights from comparative genomics.
2017 - Environ. Microbiol., 12: 4939-4952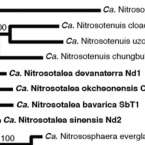
Abstract:
Obligate acidophilic members of the thaumarchaeotal genus Candidatus Nitrosotalea play an important role in nitrification in acidic soils, but their evolutionary and physiological adaptations to acidic environments are still poorly understood, with only a single member of this genus (Ca. N. devanaterra) having its genome sequenced. In this study, we sequenced the genomes of two additional cultured Ca. Nitrosotalea strains, extracted an almost complete Ca. Nitrosotalea metagenome-assembled genome from an acidic fen, and performed comparative genomics of the four Ca. Nitrosotalea genomes with 19 other archaeal ammonia oxidiser genomes. Average nucleotide and amino acid identities revealed that the four Ca. Nitrosotalea strains represent separate species within the genus. The four Ca. Nitrosotalea genomes contained a core set of 103 orthologous gene families absent from all other ammonia-oxidizing archaea and, for most of these gene families, expression could be demonstrated in laboratory culture or the environment via proteomic or metatranscriptomic analyses respectively. Phylogenetic analyses indicated that four of these core gene families were acquired by the Ca. Nitrosotalea common ancestor via horizontal gene transfer from acidophilic representatives of Euryarchaeota. We hypothesize that gene exchange with these acidophiles contributed to the competitive success of the Ca. Nitrosotalea lineage in acidic environments.
Abiotic Conversion of Extracellular NH2OH Contributes to N2O Emission during Ammonia Oxidation.
2017 - Environ. Sci. Technol., 22: 13122-13132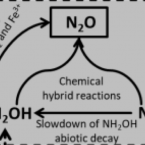
Abstract:
Abiotic processes involving the reactive ammonia-oxidation intermediates nitric oxide (NO) or hydroxylamine (NH2OH) for N2O production have been indicated recently. The latter process would require the availability of substantial amounts of free NH2OH for chemical reactions during ammonia (NH3) oxidation, but little is known about extracellular NH2OH formation by the different clades of ammonia-oxidizing microbes. Here we determined extracellular NH2OH concentrations in culture media of several ammonia-oxidizing bacteria (AOB) and archaea (AOA), as well as one complete ammonia oxidizer (comammox) enrichment (Ca. Nitrospira inopinata) during incubation under standard cultivation conditions. NH2OH was measurable in the incubation media of Nitrosomonas europaea, Nitrosospira multiformis, Nitrososphaera gargensis, and Ca. Nitrosotenuis uzonensis, but not in media of the other tested AOB and AOA. NH2OH was also formed by the comammox enrichment during NH3 oxidation. This enrichment exhibited the largest NH2OH:final product ratio (1.92%), followed by N. multiformis (0.56%) and N. gargensis (0.46%). The maximum proportions of NH4+ converted to N2O via extracellular NH2OH during incubation, estimated on the basis of NH2OH abiotic conversion rates, were 0.12%, 0.08%, and 0.14% for AOB, AOA, and Ca. Nitrospira inopinata, respectively, and were consistent with published NH4+:N2O conversion ratios for AOB and AOA.
Bottled aqua incognita: Microbiota assembly and dissolved organic matter diversity in natural mineral waters
2017 - Microbiome, 5: 126
Abstract:
Background: Non-carbonated natural mineral waters contain microorganisms that regularly grow after bottling despite low concentrations of dissolved organic matter (DOM). Yet, the compositions of bottled water microbiota and organic substrates that fuel microbial activity, and how both change after bottling, are still largely unknown.
Results: We performed a multifaceted analysis of microbiota and DOM diversity in twelve natural mineral waters from six European countries. 16S rRNA gene-based analyses showed that less than ten species-level operational taxonomic units (OTUs) dominated the bacterial communities in the water phase and associated with the bottle wall after a short phase of post-bottling growth. Members of the betaproteobacterial genera Curvibacter, Aquabacterium, and Polaromonas (Comamonadaceae) grew in most waters and represent ubiquitous, mesophilic, heterotrophic aerobes in bottled waters. Ultrahigh-resolution mass spectrometry of DOM in bottled waters and their corresponding source waters identified thousands of molecular formulae characteristic of mostly refractory, soil-derived DOM.
Conclusions. The bottle environment, including source water physicochemistry, selected for growth of a similar low-diversity microbiota across various bottled waters. Relative abundance changes of hundreds of multi-carbon molecules were related to growth of less than ten abundant OTUs. We thus speculate that individual bacteria cope with oligotrophic conditions by simultaneously consuming diverse DOM molecules.
Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay
2017 - Appl Environ Microbiol, 83: e01547-17
Abstract:
Most sulfate-reducing microorganisms (SRM) present in subsurface marine sediments belong to uncultured groups only distantly related to known SRM and it remains unclear how changing geochemical zones and sediment depth influence their community structure. We mapped the community composition and abundance of SRM by amplicon-sequencing and quantifying dsrB, which encodes dissimilatory sulfite reductase subunit beta, in sediment samples covering different vertical geochemical zones ranging from the surface sediment to the deep sulfate-depleted subsurface at four locations in Aarhus Bay, Denmark. SRM were present in all geochemical zones including sulfate-depleted methanogenic sediment. The biggest shift in SRM community composition and abundance occurring across the transition from bioturbated surface sediments into non-bioturbated sediments below, where redox fluctuations and input of fresh organic matter due to macrofaunal activity are absent. SRM abundance correlated with sulfate reduction rates determined for the same sediments. Sulfate availability showed weaker correlation with SRM abundances and no significant correlation with the composition of the SRM community. The overall SRM species diversity decreased with depth, yet we identified a subset of highly abundant community members that persists across all vertical geochemical zones of all stations. We conclude that subsurface SRM communities assemble by persistence of members of the surface community and that the transition from the bioturbated surface sediment to the unmixed sediment below is a main site of assembly of the subsurface SRM community.
Kinetic analysis of a complete nitrifier reveals an oligotrophic lifestyle.
2017 - Nature, 549: 269-272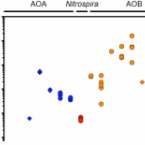
Abstract:
Nitrification, the oxidation of ammonia (NH3) via nitrite (NO2(-)) to nitrate (NO3(-)), is a key process of the biogeochemical nitrogen cycle. For decades, ammonia and nitrite oxidation were thought to be separately catalysed by ammonia-oxidizing bacteria (AOB) and archaea (AOA), and by nitrite-oxidizing bacteria (NOB). The recent discovery of complete ammonia oxidizers (comammox) in the NOB genus Nitrospira, which alone convert ammonia to nitrate, raised questions about the ecological niches in which comammox Nitrospira successfully compete with canonical nitrifiers. Here we isolate a pure culture of a comammox bacterium, Nitrospira inopinata, and show that it is adapted to slow growth in oligotrophic and dynamic habitats on the basis of a high affinity for ammonia, low maximum rate of ammonia oxidation, high growth yield compared to canonical nitrifiers, and genomic potential for alternative metabolisms. The nitrification kinetics of four AOA from soil and hot springs were determined for comparison. Their surprisingly poor substrate affinities and lower growth yields reveal that, in contrast to earlier assumptions, AOA are not necessarily the most competitive ammonia oxidizers present in strongly oligotrophic environments and that N. inopinata has the highest substrate affinity of all analysed ammonia oxidizer isolates except the marine AOA Nitrosopumilus maritimus SCM1 (ref. 3). These results suggest a role for comammox organisms in nitrification under oligotrophic and dynamic conditions.
In situ architecture, function, and evolution of a contractile injection system.
2017 - Science, 6352: 713-717
Abstract:
Contractile injection systems mediate bacterial cell-cell interactions by a bacteriophage tail-like structure. In contrast to extracellular systems, the type 6 secretion system (T6SS) is defined by intracellular localization and attachment to the cytoplasmic membrane. Here we used cryo-focused ion beam milling, electron cryotomography, and functional assays to study a T6SS in Amoebophilus asiaticus The in situ architecture revealed three modules, including a contractile sheath-tube, a baseplate, and an anchor. All modules showed conformational changes upon firing. Lateral baseplate interactions coordinated T6SSs in hexagonal arrays. The system mediated interactions with host membranes and may participate in phagosome escape. Evolutionary sequence analyses predicted that T6SSs are more widespread than previously thought. Our insights form the basis for understanding T6SS key concepts and exploring T6SS diversity.
Identification of secondary metabolite gene clusters in the Pseudovibrio genus reveals encouraging biosynthetic potential toward the production of novel bioactive compounds
2017 - Front Microbiol, 8: 1494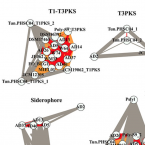
Abstract:
Increased incidences of antimicrobial resistance and the emergence of pan-resistant 'superbugs' have provoked an extreme sense of urgency amongst researchers focusing on the discovery of potentially novel antimicrobial compounds. A strategic shift in focus from the terrestrial to the marine environment has resulted in the discovery of a wide variety of structurally and functionally diverse bioactive compounds from numerous marine sources, including sponges. Bacteria found in close association with sponges and other marine invertebrates have recently gained much attention as potential sources of many of these novel bioactive compounds. Members of the genus Pseudovibrio are one such group of organisms. In this study, we interrogate the genomes of 21 Pseudovibrio strains isolated from a variety of marine sources, for the presence, diversity and distribution of biosynthetic gene clusters (BGCs). We expand on results obtained from antiSMASH analysis to demonstrate the similarity between the Pseudovibrio-related BGCs and those characterized in other bacteria and corroborate our findings with phylogenetic analysis. We assess how domain organization of the most abundant type of BGCs present among the isolates (Non-ribosomal peptide synthetases and Polyketide synthases) may influence the diversity of compounds produced by these organisms and highlight for the first time the potential for novel compound production from this genus of bacteria, using a genome guided approach.
AmoA-targeted polymerase chain reaction primers for the specific detection and quantification of comammox Nitrospira in the environment
2017 - Front Microbiol, 8:1508
Abstract:
Nitrification, the oxidation of ammonia via nitrite to nitrate, has always been considered to be catalyzed by the concerted activity of ammonia- and nitrite-oxidizing microorganisms. Only recently, complete ammonia oxidizers (‘comammox’), which oxidize ammonia to nitrate on their own, were identified in the bacterial genus Nitrospira, previously assumed to contain only canonical nitrite oxidizers. Nitrospira are widespread in nature, but for assessments of the distribution and functional importance of comammox Nitrospira in ecosystems, cultivation-independent tools to distinguish comammox from strictly nitrite oxidizing Nitrospira are required. Here we developed new PCR primer sets that specifically target the amoA genes coding for subunit A of the distinct ammonia monooxygenase of comammox Nitrospira. While existing primers capture only a fraction of the known comammox amoA diversity, the new primer sets cover as much as 95% of the comammox amoA clade A and 92% of the clade B sequences in a reference database containing 326 comammox amoA genes with sequence information at the primer binding sites. Application of the primers to 13 samples from engineered systems (a groundwater well, drinking water treatment and wastewater treatment plants) and other habitats (rice paddy and forest soils, rice rhizosphere, brackish lake sediment and freshwater biofilm) detected comammox Nitrospira in all samples and revealed a considerable diversity of comammox in most habitats. Excellent primer specificity for comammox amoA was achieved by avoiding the use of highly degenerate primer preparations and by using equimolar mixtures of oligonucleotides that match existing comammox amoA genes. Quantitative PCR with these equimolar primer mixtures was highly sensitive and specific, and enabled the efficient quantification of clade A and clade B comammox amoA gene copy numbers in environmental samples. The measured relative abundances of comammox Nitrospira, compared to canonical ammonia oxidizers, were highly variable across environments. The new comammox amoA-targeted primers enable more encompassing future studies of nitrifying microorganisms in diverse habitats. For example, they may be used to monitor the population dynamics of uncultured comammox organisms under changing environmental conditions and in response to altered treatments in engineered and agricultural ecosystems.
Unexpected genomic features in widespread intracellular bacteria: evidence for motility of marine chlamydiae.
2017 - ISME J, 10: 2334-2344
Abstract:
Chlamydiae are obligate intracellular bacteria comprising important human pathogens and symbionts of protists. Molecular evidence indicates a tremendous diversity of chlamydiae particularly in marine environments, yet our current knowledge is based mainly on terrestrial representatives. Here we provide first insights into the biology of marine chlamydiae representing three divergent clades. Our analysis of single-cell amplified genomes revealed hallmarks of the chlamydial lifestyle, supporting the ancient origin of their characteristic developmental cycle and major virulence mechanisms. Surprisingly, these chlamydial genomes encode a complete flagellar apparatus, a previously unreported feature. We show that flagella are an ancient trait that was subject to differential gene loss among extant chlamydiae. Together with a chemotaxis system, these marine chlamydiae are likely motile, with flagella potentially playing a role during host cell infection. This study broadens our view on chlamydial biology and indicates a largely underestimated potential to adapt to different hosts and environments.
'Candidatus Cochliophilus cryoturris' (Coxiellaceae), a symbiont of the testate amoeba Cochliopodium minus.
2017 - Sci Rep, 1: 3394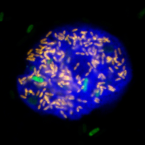
Abstract:
Free-living amoebae are well known for their role in controlling microbial community composition through grazing, but some groups, namely Acanthamoeba species, also frequently serve as hosts for bacterial symbionts. Here we report the first identification of a bacterial symbiont in the testate amoeba Cochliopodium. The amoeba was isolated from a cooling tower water sample and identified as C. minus. Fluorescence in situ hybridization and transmission electron microscopy revealed intracellular symbionts located in vacuoles. 16S rRNA-based phylogenetic analysis identified the endosymbiont as member of a monophyletic group within the family Coxiellaceae (Gammaprotebacteria; Legionellales), only moderately related to known amoeba symbionts. We propose to tentatively classify these bacteria as 'Candidatus Cochliophilus cryoturris'. Our findings add both, a novel group of amoeba and a novel group of symbionts, to the growing list of bacteria-amoeba relationships.
Evaluating the Detection of Hydrocarbon-Degrading Bacteria in 16S rRNA Gene Sequencing Surveys.
2017 - Front Microbiol, 8: 2460
Abstract:
Hydrocarbonoclastic bacteria (HCB) play a key role in the biodegradation of oil hydrocarbons in marine and other environments. A small number of taxa have been identified as obligate HCB, notably the Gammaproteobacterial genera Alcanivorax, Cycloclasticus, Marinobacter, Neptumonas, Oleiphilus, Oleispira, and Thalassolituus, as well as the Alphaproteobacterial genus Thalassospira. Detection of HCB in amplicon-based sequencing surveys relies on high coverage by PCR primers and accurate taxonomic classification. In this study, we performed a phylogenetic analysis to identify 16S rRNA gene sequence regions that represent the breadth of sequence diversity within these taxa. Using validated sequences, we evaluated 449 universal 16S rRNA gene-targeted bacterial PCR primer pairs for their coverage of these taxa. The results of this analysis provide a practical framework for selection of suitable primer sets for optimal detection of HCB in sequencing surveys.
Vibrational Spectroscopy for Imaging Single Microbial Cells in Complex Biological Samples.
2017 - Front Microbiol, 8: 675
Abstract:
Vibrational spectroscopy is increasingly used for the rapid and non-destructive imaging of environmental and medical samples. Both Raman and Fourier-transform infrared (FT-IR) imaging have been applied to obtain detailed information on the chemical composition of biological materials, ranging from single microbial cells to tissues. Due to its compatibility with methods such as stable isotope labeling for the monitoring of cellular activities, vibrational spectroscopy also holds considerable power as a tool in microbial ecology. Chemical imaging of undisturbed biological systems (such as live cells in their native habitats) presents unique challenges due to the physical and chemical complexity of the samples, potential for spectral interference, and frequent need for real-time measurements. This Mini Review provides a critical synthesis of recent applications of Raman and FT-IR spectroscopy for characterizing complex biological samples, with a focus on developments in single-cell imaging. We also discuss how new spectroscopic methods could be used to overcome current limitations of single-cell analyses. Given the inherent complementarity of Raman and FT-IR spectroscopic methods, we discuss how combining these approaches could enable us to obtain new insights into biological activities either in situ or under conditions that simulate selected properties of the natural environment.
Members of the Oral Microbiota Are Associated with IL-8 Release by Gingival Epithelial Cells in Healthy Individuals.
2017 - Front Microbiol, 8: 416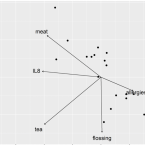
Abstract:
The triggers for the onset of oral diseases are still poorly understood. The aim of this study was to characterize the oral bacterial community in healthy humans and its association with nutrition, oral hygiene habits, and the release of the inflammatory marker IL-8 from gingival epithelial cells (GECs) with and without stimulation by bacterial endotoxins to identify possible indicator operational taxonomic units (OTUs) associated with inflammatory marker status. GECs from 21 healthy participants (13 females, 8 males) were incubated with or without addition of bacterial lipopolysaccharides (LPSs), and the oral microbiota was profiled using 16S rRNA gene-targeted sequencing. The basal IL-8 release after 6 h was between 9.9 and 98.2 pg/ml, and bacterial communities were characteristic for healthy oral microbiota. The composition of the oral microbiota was associated with basal IL-8 levels, the intake of meat, tea, white wine, sweets and the use of chewing gum, as well as flossing habits, allergies, gender and body mass index. Additionally, eight OTUs were associated with high basal levels of IL-8 and GEC response to LPS, with high basal levels of IL-8, and 1 with low basal levels of IL8. The identification of indicator bacteria in healthy subjects with high levels of IL-8 release is of importance as they may be promising early warning indicators for the possible onset of oral diseases.
Biphasic Metabolism and Host Interaction of a Chlamydial Symbiont.
2017 - mSystems, 2: e00202-16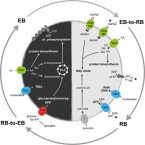
Abstract:
Chlamydiae are obligate intracellular bacteria comprising well-known human pathogens and ubiquitous symbionts of protists, which are characterized by a unique developmental cycle. Here we comprehensively analyzed gene expression dynamics of Protochlamydia amoebophila during infection of its Acanthamoeba host by RNA sequencing. This revealed a highly dynamic transcriptional landscape, where major transcriptional shifts are conserved among chlamydial symbionts and pathogens. Our data served to propose a time-resolved model for type III protein secretion during the developmental cycle, and we provide evidence for a biphasic metabolism of P. amoebophila during infection, which involves energy parasitism and amino acids as the carbon source during initial stages and a postreplicative switch to endogenous glucose-based ATP production. This fits well with major transcriptional changes in the amoeba host, where upregulation of complex sugar breakdown precedes the P. amoebophila metabolic switch. The biphasic chlamydial metabolism represents a unique adaptation to exploit eukaryotic host cells, which likely contributed to the evolutionary success of this group of microbes. IMPORTANCE Chlamydiae are known as major bacterial pathogens of humans, causing the ancient disease trachoma, but they are also frequently found in the environment where they infect ubiquitous protists such as amoebae. All known chlamydiae require a eukaryotic host cell to thrive. Using the environmental chlamydia Protochlamydia amoebophila within its natural host, Acanthamoeba castellanii, we investigated gene expression dynamics in vivo and throughout the complete chlamydial developmental cycle for the first time. This allowed us to infer how a major virulence mechanism, the type III secretion system, is regulated and employed, and we show that the physiology of chlamydiae undergoes a complete shift regarding carbon metabolism and energy generation. This study provides comprehensive insights into the infection strategy of chlamydiae and reveals a unique adaptation to life within a eukaryotic host cell.
HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis
2017 - Cancer Res., 77: 2424-2438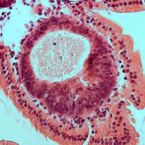
Abstract:
HuR is an RNA-binding protein implicated in immune homeostasis and various cancers, including colorectal cancer. HuR binding to AU-rich elements within the 3' untranslated region of mRNAs encoding oncogenes, growth factors, and various cytokines leads message stability and translation. In this study, we evaluated HuR as a small-molecule target for preventing colorectal cancer in high-risk groups such as those with familial adenomatosis polyposis (FAP) or inflammatory bowel disease (IBD). In human specimens, levels of cytoplasmic HuR were increased in colonic epithelial cells from patients with IBD, IBD-cancer, FAP-adenoma, and colorectal cancer, but not in patients with IBD-dysplasia. Intraperitoneal injection of the HuR small-molecule inhibitor MS-444 in AOM/DSS mice, a model of IBD and inflammatory colon cancer, augmented DSS-induced weight loss and increased tumor multiplicity, size, and invasiveness. MS-444 treatment also abrogated tumor cell apoptosis and depleted tumor-associated eosinophils, accompanied by a decrease in IL18 and eotaxin-1. In contrast, HuR inhibition in APCMin mice, a model of FAP and colon cancer, diminished the number of small intestinal tumors generated. In this setting, fecal microbiota, evaluated by 16S rRNA gene amplicon sequencing, shifted to a state of reduced bacterial diversity, with an increased representation of Prevotella, Akkermansia, and Lachnospiraceae Taken together, our results indicate that HuR activation is an early event in FAP-adenoma but is not present in IBD-dysplasia. Furthermore, our results offer a preclinical proof of concept for HuR inhibition as an effective means of FAP chemoprevention, with caution advised in the setting of IBD.
Giant viruses with an expanded complement of translation system components.
2017 - Science, 6333: 82-85
Abstract:
The discovery of giant viruses blurred the sharp division between viruses and cellular life. Giant virus genomes encode proteins considered as signatures of cellular organisms, particularly translation system components, prompting hypotheses that these viruses derived from a fourth domain of cellular life. Here we report the discovery of a group of giant viruses (Klosneuviruses) in metagenomic data. Compared with other giant viruses, the Klosneuviruses encode an expanded translation machinery, including aminoacyl transfer RNA synthetases with specificities for all 20 amino acids. Notwithstanding the prevalence of translation system components, comprehensive phylogenomic analysis of these genes indicates that Klosneuviruses did not evolve from a cellular ancestor but rather are derived from a much smaller virus through extensive gain of host genes.
Crenothrix are major methane consumers in stratified lakes.
2017 - ISME J, 9: 2124-2140
Abstract:
Methane-oxidizing bacteria represent a major biological sink for methane and are thus Earth's natural protection against this potent greenhouse gas. Here we show that in two stratified freshwater lakes a substantial part of upward-diffusing methane was oxidized by filamentous gamma-proteobacteria related to Crenothrix polyspora. These filamentous bacteria have been known as contaminants of drinking water supplies since 1870, but their role in the environmental methane removal has remained unclear. While oxidizing methane, these organisms were assigned an 'unusual' methane monooxygenase (MMO), which was only distantly related to 'classical' MMO of gamma-proteobacterial methanotrophs. We now correct this assignment and show that Crenothrix encode a typical gamma-proteobacterial PmoA. Stable isotope labeling in combination swith single-cell imaging mass spectrometry revealed methane-dependent growth of the lacustrine Crenothrix with oxygen as well as under oxygen-deficient conditions. Crenothrix genomes encoded pathways for the respiration of oxygen as well as for the reduction of nitrate to N2O. The observed abundance and planktonic growth of Crenothrix suggest that these methanotrophs can act as a relevant biological sink for methane in stratified lakes and should be considered in the context of environmental removal of methane.
Variant profiling of evolving prokaryotic populations.
2017 - PeerJ, e2997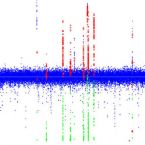
Abstract:
Genomic heterogeneity of bacterial species is observed and studied in experimental evolution experiments and clinical diagnostics, and occurs as micro-diversity of natural habitats. The challenge for genome research is to accurately capture this heterogeneity with the currently used short sequencing reads. Recent advances in NGS technologies improved the speed and coverage and thus allowed for deep sequencing of bacterial populations. This facilitates the quantitative assessment of genomic heterogeneity, including low frequency alleles or haplotypes. However, false positive variant predictions due to sequencing errors and mapping artifacts of short reads need to be prevented. We therefore created VarCap, a workflow for the reliable prediction of different types of variants even at low frequencies. In order to predict SNPs, InDels and structural variations, we evaluated the sensitivity and accuracy of different software tools using synthetic read data. The results suggested that the best sensitivity could be reached by a union of different tools, however at the price of increased false positives. We identified possible reasons for false predictions and used this knowledge to improve the accuracy by post-filtering the predicted variants according to properties such as frequency, coverage, genomic environment/localization and co-localization with other variants. We observed that best precision was achieved by using an intersection of at least two tools per variant. This resulted in the reliable prediction of variants above a minimum relative abundance of 2%. VarCap is designed for being routinely used within experimental evolution experiments or for clinical diagnostics. The detected variants are reported as frequencies within a VCF file and as a graphical overview of the distribution of the different variant/allele/haplotype frequencies. The source code of VarCap is available at https://github.com/ma2o/VarCap. In order to provide this workflow to a broad community, we implemeted VarCap on a Galaxy webserver, which is accessible at http://galaxy.csb.univie.ac.at.
Capturing the genetic makeup of the active microbiome in situ.
2017 - ISME J, 9: 1949-1963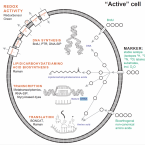
Abstract:
More than any other technology, nucleic acid sequencing has enabled microbial ecology studies to be complemented with the data volumes necessary to capture the extent of microbial diversity and dynamics in a wide range of environments. In order to truly understand and predict environmental processes, however, the distinction between active, inactive and dead microbial cells is critical. Also, experimental designs need to be sensitive toward varying population complexity and activity, and temporal as well as spatial scales of process rates. There are a number of approaches, including single-cell techniques, which were designed to study in situ microbial activity and that have been successively coupled to nucleic acid sequencing. The exciting new discoveries regarding in situ microbial activity provide evidence that future microbial ecology studies will indispensably rely on techniques that specifically capture members of the microbiome active in the environment. Herein, we review those currently used activity-based approaches that can be directly linked to shotgun nucleic acid sequencing, evaluate their relevance to ecology studies, and discuss future directions.
A 12-week intervention with nonivamide, a TRPV1 agonist, prevents a dietary-induced body fat gain and increases peripheral serotonin in moderately overweight subjects.
2017 - Mol Nutr Food Res, 5: 1600731
Abstract:
A bolus administration of 0.15 mg nonivamide has previously been demonstrated to reduce energy intake in moderately overweight men. This 12-week intervention investigated whether a daily consumption of nonivamide in a protein-based product formulation promotes a reduction in body weight in healthy overweight subjects and affects outcome measures associated with mechanisms regulating food intake, e.g. plasma concentrations of (an)orexigenic hormones, energy substrates as well as changes in fecal microbiota.
Nineteen overweight subjects were randomly assigned to either a control (C) or a nonivamide (NV) group. Changes in the body composition and plasma concentrations of satiating hormones were determined at fasting and 15, 30, 60, 90, and 120 min after a glucose load. Participants were instructed to consume 0.15 mg nonivamide per day in 450 mL of a milk shake additionally to their habitual diet. After treatment, a group difference in body fat mass change (-0.61 ± 0.36% in NV and +1.36 ± 0.38% in C) and an increase in postprandial plasma serotonin were demonstrated. Plasma metabolome and fecal microbiome read outs were not affected.
A daily intake of 0.15 mg nonivamide helps to support to maintain a healthy body composition.Microbial nutrient niches in the gut.
2017 - Environ. Microbiol., 4: 1366-1378
Abstract:
The composition and function of the mammalian gut microbiota has been the subject of much research in recent years, but the principles underlying the assembly and structure of this complex community remain incompletely understood. Processes that shape the gut microbiota are thought to be mostly niche-driven, with environmental factors such as the composition of available nutrients largely determining whether or not an organism can establish. The concept that the nutrient landscape dictates which organisms can successfully colonize and persist in the gut was first proposed in Rolf Freter's nutrient niche theory. In a situation where nutrients are perfectly mixed and there is balanced microbial growth, Freter postulated that an organism can only survive if it is able to utilize one or a few limiting nutrients more efficiently than its competitors. Recent experimental work indicates, however, that nutrients in the gut vary in space and time. We propose that in such a scenario, Freter's nutrient niche theory must be expanded to account for the co-existence of microorganisms utilizing the same nutrients but in distinct sites or at different times, and that metabolic flexibility and mixed-substrate utilization are common strategies for survival in the face of ever-present nutrient fluctuations.
Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota
2017 - mSystems, 2: e00171-16
Abstract:
Mucispirillum schaedleri is an abundant inhabitant of the intestinal mucus layer of rodents and other animals and has been suggested to be a pathobiont, a commensal that plays a role in disease. In order to gain insights into its lifestyle, we analyzed the genome and transcriptome of M. schaedleri ASF 457 and performed physiological experiments to test traits predicted by its genome. Although described as a mucus inhabitant, M. schaedleri has limited capacity for degrading host-derived mucosal glycans and other complex polysaccharides. Additionally, M. schaedleri reduces nitrate and expresses systems for scavenging oxygen and reactive oxygen species in vivo, which may account for its localization close to the mucosal tissue and expansion during inflammation. Also of note, M. schaedleri harbors a type VI secretion system and putative effector proteins and can modify gene expression in mucosal tissue, suggesting intimate interactions with its host and a possible role in inflammation. The M. schaedleri genome has been shaped by extensive horizontal gene transfer, primarily from intestinal Epsilon- and Deltaproteobacteria, indicating that horizontal gene transfer has played a key role in defining its niche in the gut ecosystem.
Astrobiology as a framework for investigating antibiotic susceptibility: a study of Halomonas hydrothermalis
2017 - J R Soc Interface, 126: online only
Abstract:
Physical and chemical boundaries for microbial multiplication on Earth are strongly influenced by interactions between environmental extremes. However, little is known about how interactions between multiple stress parameters affect the sensitivity of microorganisms to antibiotics. Here, we assessed how 12 distinct permutations of salinity, availability of an essential nutrient (iron) and atmospheric composition (aerobic or microaerobic) affect the susceptibility of a polyextremotolerant bacterium, Halomonas hydrothermalis, to ampicillin, kanamycin and ofloxacin. While salinity had a significant impact on sensitivity to all three antibiotics (as shown by turbidimetric analyses), the nature of this impact was modified by iron availability and the ambient gas composition, with differing effects observed for each compound. These two parameters were found to be of particular importance when considered in combination and, in the case of ampicillin, had a stronger combined influence on antibiotic tolerance than salinity. Our data show how investigating microbial responses to multiple extremes, which are more representative of natural habitats than single extremes, can improve our understanding of the effects of antimicrobial compounds and suggest how studies of habitability, motivated by the desire to map the limits of life, can be used to systematically assess the effectiveness of antibiotics.
Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments.
2017 - ISME J, 5: 1276-1281
Abstract:
To date, very little is known about the bacterial core community of marine sediments. Here we study the environmental distribution, abundance and ecogenomics of the gammaproteobacterial Woeseiaceae/JTB255 marine benthic group. A meta-analysis of published work shows that the Woeseiaceae/JTB255 are ubiquitous and consistently rank among the most abundant 16S rRNA gene sequences in diverse marine sediments. They account for up to 22% of bacterial amplicons and 6% of total cell counts in European and Australian coastal sediments. The analysis of a single-cell genome, metagenomic bins and the genome of the next cultured relative Woeseia oceani indicated a broad physiological range, including heterotrophy and facultative autotrophy. All tested (meta)genomes encode a truncated denitrification pathway to nitrous oxide. The broad range of energy-yielding metabolisms possibly explains the ubiquity and high abundance of Woeseiaceae/JTB255 in marine sediments, where they carry out diverse, but yet unknown ecological functions.
Happens in the best of subfamilies: establishment and repeated replacements of co-obligate secondary endosymbionts within Lachninae aphids.
2017 - Environ. Microbiol., 1: 393-408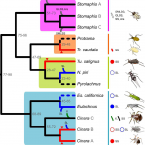
Abstract:
Virtually all aphids maintain an obligate mutualistic symbiosis with bacteria from the Buchnera genus, which produce essential nutrients for their aphid hosts. Most aphids from the Lachninae subfamily have been consistently found to house additional endosymbionts, mainly Serratia symbiotica. This apparent dependence on secondary endosymbionts was proposed to have been triggered by the loss of the riboflavin biosynthetic capability by Buchnera in the Lachninae last common ancestor. However, an integral large-scale analysis of secondary endosymbionts in the Lachninae is still missing, hampering the interpretation of the evolutionary and genomic analyses of these endosymbionts. Here, we analysed the endosymbionts of selected representatives from seven different Lachninae genera and nineteen species, spanning four tribes, both by FISH (exploring the symbionts' morphology and tissue tropism) and 16S rRNA gene sequencing. We demonstrate that all analysed aphids possess dual symbiotic systems, and while most harbour S. symbiotica, some have undergone symbiont replacement by other phylogenetically-distinct bacterial taxa. We found that these secondary associates display contrasting cell shapes and tissue tropism, and some appear to be lineage-specific. We propose a scenario for symbiont establishment in the Lachninae, followed by changes in the symbiont's tissue tropism and symbiont replacement events, thereby highlighting the extraordinary versatility of host-symbiont interactions.
Cultivation and characterization of Candidatus Nitrosocosmicus exaquare, an ammonia-oxidizing archaeon from a municipal wastewater treatment system.
2017 - ISME J, 5: 1142-1157
Abstract:
Thaumarchaeota have been detected in several industrial and municipal wastewater treatment plants (WWTPs), despite the fact that ammonia-oxidizing archaea (AOA) are thought to be adapted to low ammonia environments. However, the activity, physiology and metabolism of WWTP-associated AOA remain poorly understood. We report the cultivation and complete genome sequence of Candidatus Nitrosocosmicus exaquare, a novel AOA representative from a municipal WWTP in Guelph, Ontario (Canada). In enrichment culture, Ca. N. exaquare oxidizes ammonia to nitrite stoichiometrically, is mesophilic, and tolerates at least 15 mm of ammonium chloride or sodium nitrite. Microautoradiography (MAR) for enrichment cultures demonstrates that Ca. N. exaquare assimilates bicarbonate in association with ammonia oxidation. However, despite using inorganic carbon, the ammonia-oxidizing activity of Ca. N. exaquare is greatly stimulated in enrichment culture by the addition of organic compounds, especially malate and succinate. Ca. N. exaquare cells are coccoid with a diameter of ~1-2 μm. Phylogenetically, Ca. N. exaquare belongs to the Nitrososphaera sister cluster within the Group I.1b Thaumarchaeota, a lineage which includes most other reported AOA sequences from municipal and industrial WWTPs. The 2.99 Mbp genome of Ca. N. exaquare encodes pathways for ammonia oxidation, bicarbonate fixation, and urea transport and breakdown. In addition, this genome encodes several key genes for dealing with oxidative stress, including peroxidase and catalase. Incubations of WWTP biofilm demonstrate partial inhibition of ammonia-oxidizing activity by 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), suggesting that Ca. N. exaquare-like AOA may contribute to nitrification in situ. However, CARD-FISH-MAR showed no incorporation of bicarbonate by detected Thaumarchaeaota, suggesting that detected AOA may incorporate non-bicarbonate carbon sources or rely on an alternative and yet unknown metabolism.
Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis.
2017 - ISME J, 11: 463–477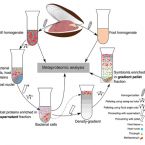
Abstract:
The hydrothermal vent mussel Bathymodiolus azoricus lives in an intimate symbiosis with two types of chemosynthetic Gammaproteobacteria in its gills: a sulfur oxidizer and a methane oxidizer. Despite numerous investigations over the last decades, the degree of interdependence between the three symbiotic partners, their individual metabolic contributions, as well as the mechanism of carbon transfer from the symbionts to the host are poorly understood. We used a combination of proteomics and genomics to investigate the physiology and metabolism of the individual symbiotic partners. Our study revealed that key metabolic functions are most likely accomplished jointly by B. azoricus and its symbionts: (1) CO2 is pre-concentrated by the host for carbon fixation by the sulfur-oxidizing symbiont, and (2) the host replenishes essential biosynthetic TCA cycle intermediates for the sulfur-oxidizing symbiont. In return (3), the sulfur oxidizer may compensate for the host's putative deficiency in amino acid and cofactor biosynthesis. We also identified numerous 'symbiosis-specific' host proteins by comparing symbiont-containing and symbiont-free host tissues and symbiont fractions. These proteins included a large complement of host digestive enzymes in the gill that are likely involved in symbiont digestion and carbon transfer from the symbionts to the host.The ISME Journal advance online publication, 1 November 2016; doi:10.1038/ismej.2016.124.
Convergent patterns in the evolution of mealybug symbioses involving different intrabacterial symbionts.
2017 - ISME J, 3: 715-726
Abstract:
Mealybugs (Insecta: Hemiptera: Pseudococcidae) maintain obligatory relationships with bacterial symbionts, which provide essential nutrients to their insect hosts. Most pseudococcinae mealybugs harbor a unique symbiosis setup with enlarged betaproteobacterial symbionts ('Candidatus Tremblaya princeps'), which themselves contain gammaproteobacterial symbionts. Here we investigated the symbiosis of the manna mealybug, Trabutina mannipara, using a metagenomic approach. Phylogenetic analyses revealed that the intrabacterial symbiont of T. mannipara represents a novel lineage within the Gammaproteobacteria, for which we propose the tentative name 'Candidatus Trabutinella endobia'. Combining our results with previous data available for the nested symbiosis of the citrus mealybug Planococcus citri, we show that synthesis of essential amino acids and vitamins and translation-related functions partition between the symbiotic partners in a highly similar manner in the two systems, despite the distinct evolutionary origin of the intrabacterial symbionts. Bacterial genes found in both mealybug genomes and complementing missing functions in both symbioses were likely integrated in ancestral mealybugs before T. mannipara and P. citri diversified. The high level of correspondence between the two mealybug systems and their highly intertwined metabolic pathways are unprecedented. Our work contributes to a better understanding of the only known intracellular symbiosis between two bacteria and suggests that the evolution of this unique symbiosis included the replacement of intrabacterial symbionts in ancestral mealybugs.
Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations
2017 - Environ. Microbiol., 1: 95-105
Abstract:
An altered gut microbiota has been linked to obesity in adulthood, although little is known about childhood obesity. The aim of this study was to characterize the composition of the gut microbiota in obese (n = 42) and normal-weight (n = 36) children aged 6 to 16. Using 16S rRNA gene-targeted sequencing, we evaluated taxa with differential abundance according to age- and sex-normalized body mass index (BMI z-score). Obesity was associated with an altered gut microbiota characterized by elevated levels of Firmicutes and depleted levels of Bacteroidetes. Correlation network analysis revealed that the gut microbiota of obese children also had increased correlation density and clustering of operational taxonomic units (OTUs). Members of the Bacteroidetes were generally better predictors of BMI z-score and obesity than Firmicutes, which was likely due to discordant responses of Firmicutes OTUs. In accordance with these observations, the main metabolites produced by gut bacteria, short chain fatty acids (SCFAs), were higher in obese children, suggesting elevated substrate utilisation. Multiple taxa were correlated with SCFA levels, reinforcing the tight link between the microbiota, SCFAs and obesity. Our results suggest that gut microbiota dysbiosis and elevated fermentation activity may be involved in the etiology of childhood obesity.
Stable isotope techniques for the assessment of host and microbiota response during gastrointestinal dysfunction
2017 - J Pediatr Gastroenterol Nutr, 64: 8-14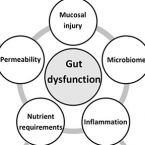
Abstract:
The International Atomic Energy Agency convened a technical meeting on environmental enteric dysfunction (EED) in Vienna (28th – 30th October 2015; https://nucleus.iaea.org/HHW/Nutrition/EED_Technical_Meeting/index.html) to bring together international experts in the fields of EED, nutrition and stable isotope technologies. Advances in stable isotope labelling techniques open up new possibilities to improve our understanding of gastrointestinal dysfunction and the role of the microbiota in host health. In the context of EED, little is known about the role gut dysfunction may play in macro- and micronutrient bioavailability and requirements and what the consequences may be for nutritional status and linear growth. Stable isotope labelling techniques have been used to assess intestinal mucosal injury and barrier function, carbohydrate digestion and fermentation, protein derived amino acid bioavailability and requirements, micronutrient bioavailability and to track microbe-microbe and microbe-host interactions at the single cell level. The non-invasive nature of stable isotope technologies potentially allows for low-hazard, field deployable tests of gut dysfunction that are applicable across all age-groups. The purpose of this review is to assess the state-of-the-art in the use of stable isotope technologies and to provide a perspective on where these technologies can be exploited to further our understanding of gut dysfunction in EED.
Book chapters and other publications
Terriglobus
2017 - in Bergey's Manual of Systematics of Archaea and Bacteria.. (Whitman WB, Rainey F, Kämpfer P, Trujillo M, Chun J, DeVos P, Hedlund B, Dedysh S)
Abstract:
Terriglobus is a genus in the phylum Acidobacteria in the family Acidobacteriaceae, order Acidobacteriales, class Acidobacteriia, subdivision 1. It currently comprises five species - Terriglobus roseus, Terriglobus saanensis, Terriglobus tenax, Terriglobus aquaticus, and Terriglobus albidus. Members of the genus are widely distributed in soils including rhizosphere soils and the phyllosphere, but is also found in freshwater and in association with insects. This genus encompasses bacteria that are chemo-organotrophs and have obligatory aerobic metabolism with an optimal growth in mildly acidic (pH ~5 to 6) and mesophilic (ca. 25 to 30°C) conditions. Colonies of Terriglobus are typically circular in form with a convex elevation and can be with or without pink pigmentation. These bacteria can use a range of different carbon sources, and nitrogen is attained by exogenous amino acids or ammonium chloride. Cells are non-motile, Gram-stain-negative with a length and width ranging from 0.8 to 2.5 µm and 0.4 to 0.9 µm, respectively. Some strains produce extracellular material, which can be visualized by microscopy or in liquid culture, generating a floc/clumping phenotype. The dominant fatty acids are iso-C15:0 and C16:1 ω7c/ C16:1 ω6c. The DNA G+C content (mol%) ranges from 57.3 to 63.2%.
The unexpected versatility of the cellulosome
2017 - Environmental Microbiology, 1: 13-14
Hidden potential: Diet-driven changes in redox level shape the rumen microbiome
2017 - Environmental Microbiology, 1: 19-20

Latest news


Latest publications
- Metabolic and phylogenetic diversity in the phylum Nitrospinota revealed by comparative genome analyses
- Viral potential to modulate microbial methane metabolism varies by habitat.
- The maternal microbiome in pregnancy, delivery, and early-stage development of neonatal microbiome after cesarean section: A prospective longitudinal study.
- Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants.
- Biomonitoring of Dietary Mycotoxin Exposure and Associated Impact on the Gut Microbiome in Nigerian Infants.
Contact
- +43 1 4277 76601
- +43 1 4277 825701
- office.dome@univie.ac.at